Evaluation of MET alteration in EGFR-mutant non-small cell lung cancer patients treated with EGFR tyrosine kinase inhibitor from paired biopsy: A retrospective cohort study
Article information

Abstract
Purpose
Mesenchymal-epithelial transition tyrosine kinase receptor (MET) amplification is one of the common acquired resistance mechanisms to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). To evaluate the usefulness of screening methods for MET status, we studied the impact of MET amplification or protein overexpression in EGFR-mutant non-small cell lung cancer patients who were treated with EGFR TKI.
Methods
A total of 214 patients treated with EGFR TKI as first-line therapy with available tissue biopsy was analyzed. Paired biopsies were obtained from 111 patients at baseline and at onset of resistance. MET status was determined by immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH).
Results
Among 111 patients with paired samples, incidence of MET alteration was increased according to both MET overexpression by IHC (14.4% to 22.5%) and MET amplification by FISH (1.8% to 8.1%) with moderated to strong IHC intensity samples after EGFR TKI treatment. In patients treated with 1st-generation EGFR TKI, MET amplification by FISH was significantly related to shorter progression-free survival (P=0.04) and overall survival (P=0.01). In contrast, there was no difference in clinical outcomes according to MET intensity of IHC. Patients harboring MET amplification by FISH were associated with poor clinical outcomes compared to those with T790M mutation at progression.
Conclusion
These results suggest that FISH is more informative than IHC for identification of patients with MET amplification as an EGFR TKI resistance mechanism. Given the poor outcome in patients who developed MET amplification, combinational trials with more active MET inhibitor are needed to overcome resistance.
INTRODUCTION
Epidermal growth factor receptor (EGFR) mutation is frequently found in non-small cell lung cancer (NSCLC) patients, especially in never-smoker, female, and Asian cohorts. EGFR tyrosine kinase inhibitors (TKIs) are the established first-line therapy in patients with NSCLC harboring EGFR mutation. However, most patients inevitably counter disease progression due to acquired resistance. T790M mutation is the most common resistance mutation and accounts for 40% to 50% of cases, followed by activation of a bypass signaling pathway such as mesenchymal-epithelial transition tyrosine kinase receptor (MET) amplification [1,2].
The MET gene and its ligand hepatocyte growth factor/scatter factor are part of a signal pathway affecting cell cycle progression, cell survival, and cell migration [2,3]. Activation of the MET gene is associated with initial and acquired resistance to EGFR TKI in NSCLC patients [4]. Amplification of the MET gene is detected in 2% to 5% of NSCLC patients, and up to 22% of patients with NSCLC who progress on first-line EGFR TKIs have MET amplification or other MET-based mechanisms of resistance [1,5]. While patients with T790M mutation have achieved prolonged overall survival (OS) after development of osimertinib, a third-generation EGFR TKI, there is no effective inhibitor for treatment of MET gene amplification [6]. Given the encouraging results of the combination of EGFR TKI with selective MET inhibitors including capmatinib, savolitinib, tepotinib, or amivantamab, it is crucial to identify patients with MET alterations as a mechanism of EGFR TKI resistance [7-10].
For identification of MET amplification, fluorescence in situ hybridization (FISH) is a commonly used technique. Usually, MET amplification is defined as a mean of five or more gene copy numbers (GCN) of MET per cell (GCN≥ 5) or a ratio of MET to centromere of chromosome 7 ratio (MET/CEP7) ≥ 2.0 [11,12]. Recently, next-generation sequencing (NGS) has been used to determine MET copy number, but the cut-off for MET amplification varies across NGS platforms [13,14]. Since MET can be overexpressed in cancers that harbor an activating genomic signature like MET amplification, MET protein overexpression using immunohistochemistry (IHC) is also a screening method for MET amplification [9]. The extent and intensity of IHC staining as assessed by a pathologist provide a semiquantitative indication of MET protein expression. In general, the degree of expression is typically quantified as a staining score on a scale of 0 to 3+, corresponding to negative (0), weak (1+), moderate (2+), or strong (3+) staining, and a staining score of 3+ in at least 50% of the cells is a commonly used cut-off for MET overexpression [15].
Given the low incidence of MET amplification and difficulty of tissue obtaining after EGFR TKI failure, it is not feasible in the clinic to screen every patient by FISH or NGS. Though MET IHC has been used as an initial screening test for MET amplification, its accuracy has been controversial [9,16,17]. Therefore, we analyzed MET amplification by FISH and protein expression by IHC in tissue biopsy samples obtained from patients at baseline and at the time of resistance to EGFR TKI to evaluate the relevance of each testing method.
METHODS
Patients
This study included 214 EGFR-mutated NSCLC biopsy specimens from Samsung Medical Center between 2015 and 2020. All patients received 1st- or 2nd-generation EGFR TKI, such as erlotinib, gefitinib, or afatinib. Biopsy samples were obtained by transbronchial lung biopsy, surgery, or percutaneous needle aspiration at baseline and/or at the time of progression. Clinical characteristics and clinical outcomes were retrospectively retrieved from electronic medical records. EGFR TKI response was evaluated by RECIST version 1.1. The study was reviewed and approved by the Institutional Review Board (IRB) of Samsung Medical Center, Seoul, Korea (IRB No. 2020-08-027). Individual consent for this retrospective analysis was waived.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue slides were stained with anti-total c-MET (SP44) rabbit monoclonal primary antibody (#7904430, Ventana Medical Systems, Tucson, AZ, USA). The MET IHC results were evaluated by a pathologist, and staining intensity of each slide was graded as negative (0), weak (1+), moderate (2+), or strong (3+). MET overexpression was defined as IHC 3+ in more than 50% of tumor cells [15].
Fluorescence in situ hybridization
For cases with moderate to strong staining, MET FISH was performed to confirm amplification using ZytoLight SPEC MET/CEN7 dual-color probe (Zytovision, Bremerhaven, Germany). Fifty nuclei of tumor cells were selected using 4′,6-diamidino-2-phenylindole (DAPI) and the average count of MET signals was determined for each specimen. MET amplification was defined as MET GCN ≥ 5 or MET/CEP7 ratio ≥ 2 [11,12].
Statistical analyses
Descriptive statistics were used to describe clinical characteristics of patients. Correlation between FISH and IHC was evaluated with Fisher’s exact test. Survival was analyzed with the Kaplan-Meier method and compared by log-rank test. Progression-free survival (PFS) was defined as the time from initiation of EGFR TKI treatment to disease progression or death from any cause, and OS was defined as the time from initiation of EGFR TKI treatment to death of any cause. Statistical significance was defined by two-sided test as P < 0.05. All analyses were performed using R software version 3.6.3 (R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism version 3 8.4.3 (GraphPad Software Inc., San Diego, CA, USA).
RESULTS
Patient demographics and clinicopathological characteristics
A total of 214 patients who had undergone biopsy either before EGFR TKI treatment (n= 183) or at progression (n= 142) was included in this study. The median age was 63 years (range, 30 to 89), 38.3% were male, all of them had adenocarcinoma, 66.4% were never-smokers, and 96.3% were stage IV. The EGFR mutation subtypes detected before EGFR TKI treatment were exon 19 deletion in 60.7%, L858R substitution in 34.1%, and others in 5.1%. The EGFR TKIs used were afatinib, gefitinib, and erlotinib in 44.9%, 42.5%, and 12.6% of patients, respectively (Table 1). Among 214 patients, 111 (51.9%) had paired biopsy samples at baseline and progression. Others had only baseline biopsy sample (n= 72, 33.6%) or only progression biopsy sample (n= 31, 14.5%).

Clinical characteristics of patients
Clinical characteristics of 111 patients who had paired biopsy samples at baseline and progression were similar to those of entire study population. The median age was 63 years (range, 30 to 89), 36.9% were male, all of them had adenocarcinoma, 68.5% were never-smokers, and 96.4% were stage IV. The EGFR mutation subtypes were exon 19 deletion in 55.9%, L858R substitution in 39.6%, and others in 4.5%. The EGFR TKIs used were afatinib, gefitinib, and erlotinib in 38.7%, 45.0%, and 16.2%, respectively (Table 1).
Prevalence of MET alteration at baseline and at progression to EGFR TKI treatment
To determine the MET expression status, MET IHC was conducted in 325 biopsy samples (baseline, 183 samples; progression, 142 samples) (Fig. 1A, Supplementary Fig. 1). As patients with high MET protein expression showed high consistency with MET amplification [16], MET FISH was further evaluated in 65 biopsy samples (baseline, 20 samples; progression, 45 samples) (Supplementary Fig. 1) which had moderate or strong IHC intensity. Positive MET overexpression (3+) by IHC was found in 16.4% (30/183) and 22.5% (32/142) of patients at baseline and progression, respectively (Fig. 1B). In 183 baseline samples, two showed MET amplification and overexpression on both FISH (GCN≥ 5) and IHC (3+). Moderate MET IHC intensity (2+) was identified in one sample by FISH (GCN = 6.5). In comparison, in 65 FISH samples with moderated to strong IHC intensity of 142 progression samples, 11 cases were positive for MET amplification and overexpression in both FISH (GCN≥ 5) and IHC (3+). There was no MET amplification by FISH in progression samples with weak (0–1+) or moderate (2+) IHC intensity.
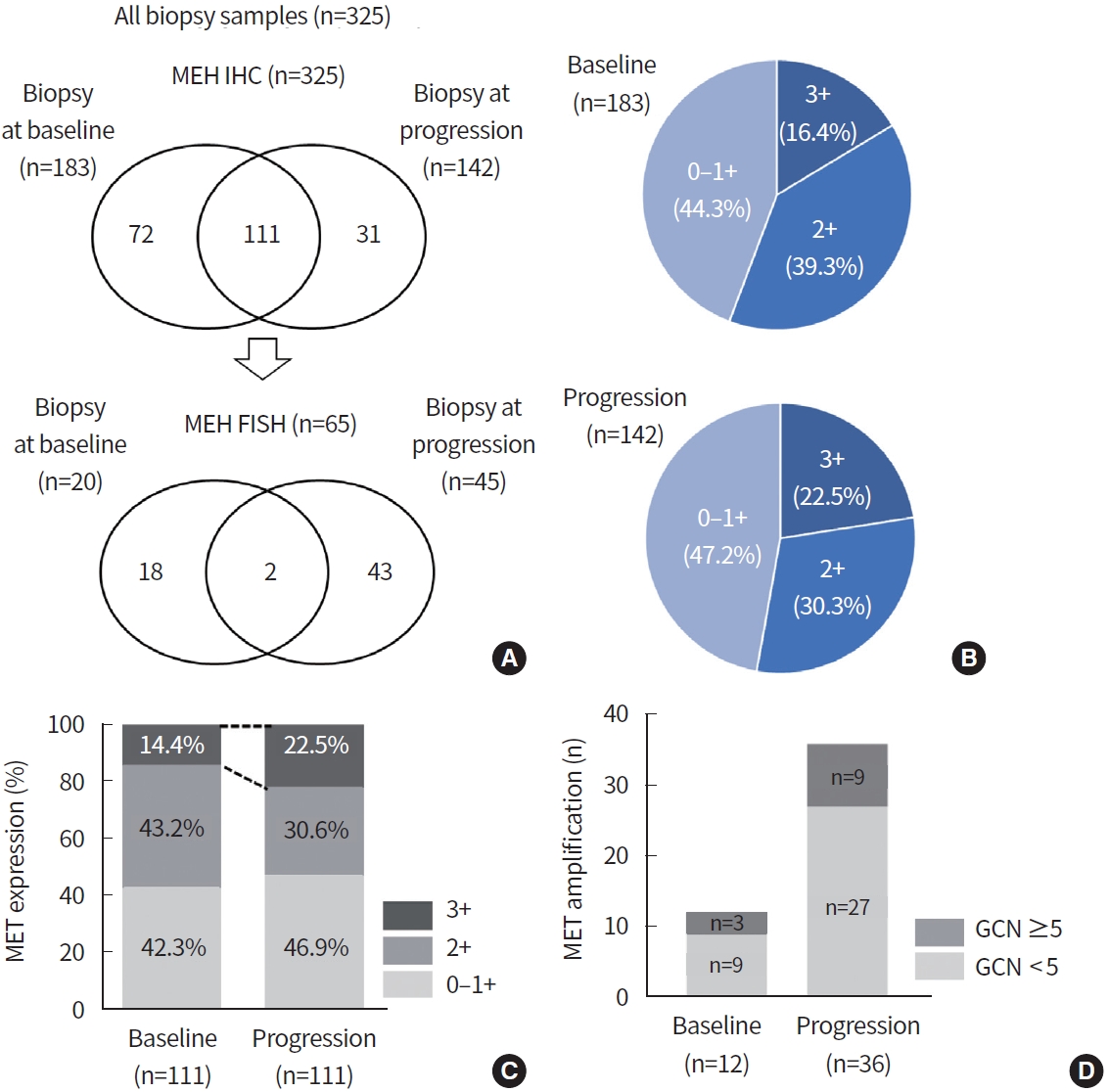
Mesenchymal-epithelial transition tyrosine kinase receptor (MET) alteration in biopsy samples before and after epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) treatment. (A) Overview of analyzed samples. (B) The relative frequencies of MET staining intensity by immunohistochemistry at baseline and progression. Among 111 paired biopsy samples, proportion of MET overexpression (C) and MET amplification (D) increased after EGFR TKI treatment compared to baseline. FISH, fluorescence in situ hybridization; GCN, gene copy number.
To verify MET alteration by EGFR TKI treatment, we analyzed only paired biopsy cases. Among 111 paired biopsy samples, MET overexpression by IHC (3+) was found in 14.4% (16/111) and 22.5% (25/111) at baseline and progression, respectively (Fig. 1C). In addition, MET amplification by FISH was identified in three patients at baseline and nine patients at progression (Fig. 1D). Proportion of MET-altered patients was increased at the time of progression according to both IHC and FISH.
Concordance of MET status between FISH and IHC
To evaluate the relevance between FISH and IHC, we compared the concordance of MET amplification by FISH and overexpression by IHC. Of the 65 samples analyzed with both IHC and FISH, 20.0% harbored MET amplification and overexpression in both tests (n= 13). The rate of MET amplification or overexpression detected only by FISH or only by IHC was 21.5% (n = 14) and 66.2% (n = 43), respectively. Compared with FISH, IHC showed a sensitivity of 92.9%, a specificity of 41.2%, a positive predictive value of 30.2%, and a negative predictive value of 95.5%; a 52.3% concordance rate was observed between the methods (Table 2).

Comparison of MET FISH and IHC (n=65)
Relationship between T790M mutation and MET amplification
Both T790M mutation status and MET amplification were available for 142 patients who underwent biopsy at progression (Table 3). The T790M mutation was found in 56.3% (80/142) of patients. Although MET overexpression by IHC (3+) was found in 11.3% (9/80) of patients in the T790M-positive group, there was no case of MET amplification by FISH (GCN ≥ 5). In contrast, among 62 patients with T790M negativity, MET overexpression by IHC (3+) was found in 37.1% (23/62) of patients and 11 patients had MET overexpression and MET amplification by both IHC and FISH. Presence of T790M mutation showed an inverse relationship with proportion of MET amplification.

Specimen characteristics of 142 progression samples
Clinical outcome in patients with MET status by FISH and IHC
To assess the prognostic impact of MET amplification status, we analyzed PFS and OS in 1st- and 2nd-generation EGFR TKI-treated groups. In the 1st-generation EGFR TKI-treated group, MET amplification was confirmed in six samples (one at baseline and five at progression). PFS and OS in MET amplification-positive patients were significantly shorter than in others. Median PFS was 8.6 and 13.7 months in MET amplification-positive patients and others, respectively (hazard ratio [HR], 2.26; 95% confidence interval [CI], 0.68 to 7.48; P= 0.04) (Fig. 2A). The median OS was 23.5 months in MET amplification-positive patients and undefined in others (HR, 3.55; 95% CI, 0.58 to 21.52; P= 0.01) (Fig. 2B). In contrast, for the 2nd-generation EGFR TKI-treated group, MET amplification was confirmed in eight samples (two at baseline and six at progression). There was no significant difference in PFS (P= 0.61) or OS (P= 0.83) between patients with or without MET amplification (Fig. 2C, D). In addition, there was no significant difference in PFS or OS according to the status of MET overexpression at the time of progression in both 1st- or 2nd-generation EGFR TKI-treated patients (Supplementary Fig. 2).

Prognostic significance of mesenchymal-epithelial transition tyrosine kinase receptor (MET) amplification by fluorescence in situ hybridization in patients treated with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). In the 1st-generation EGFR TKI group, MET amplification-positive patients showed significantly shorter (A) progression-free survival (PFS) and (B) overall survival (OS) than others. In the 2nd-generation EGFR TKI group, patient stratification by MET amplification had no effects on (C) PFS and (D) OS. HR, hazard ratio; CI, confidence interval.
Clinical outcomes according to MET amplification and T790M mutation
For the 1st-generation EGFR TKI group, the T790M mutation was found in 64 patients and MET amplification of T790Mnegative patients was confirmed in six samples (one at baseline and five at progression). PFS was significantly shorter in MET amplification-positive patients compared to T790M-positive patients. The median PFS was 8.6 and 15.5 months in MET amplification-positive and T790M-positive patients, respectively (HR, 2.59; 95% CI, 0.72 to 9.27; P= 0.02) (Fig. 3A). Also, OS was significantly shorter in MET amplification-positive patients (median, 23.5 months) compared to T790M-positive patients (median, undefined) (HR, 5.38; 95% CI, 0.65 to 44.35; P = 0.0008) (Fig. 3B). In the 2nd-generation EGFR TKI group, the T790M mutation was found in 50 patients and MET amplification of T790M-negative patients was confirmed at six progression samples. However, there was no difference in PFS or OS between MET amplification-positive and T790M-positive patients in the 2nd-generation EGFR TKI group (Fig. 3C, D).
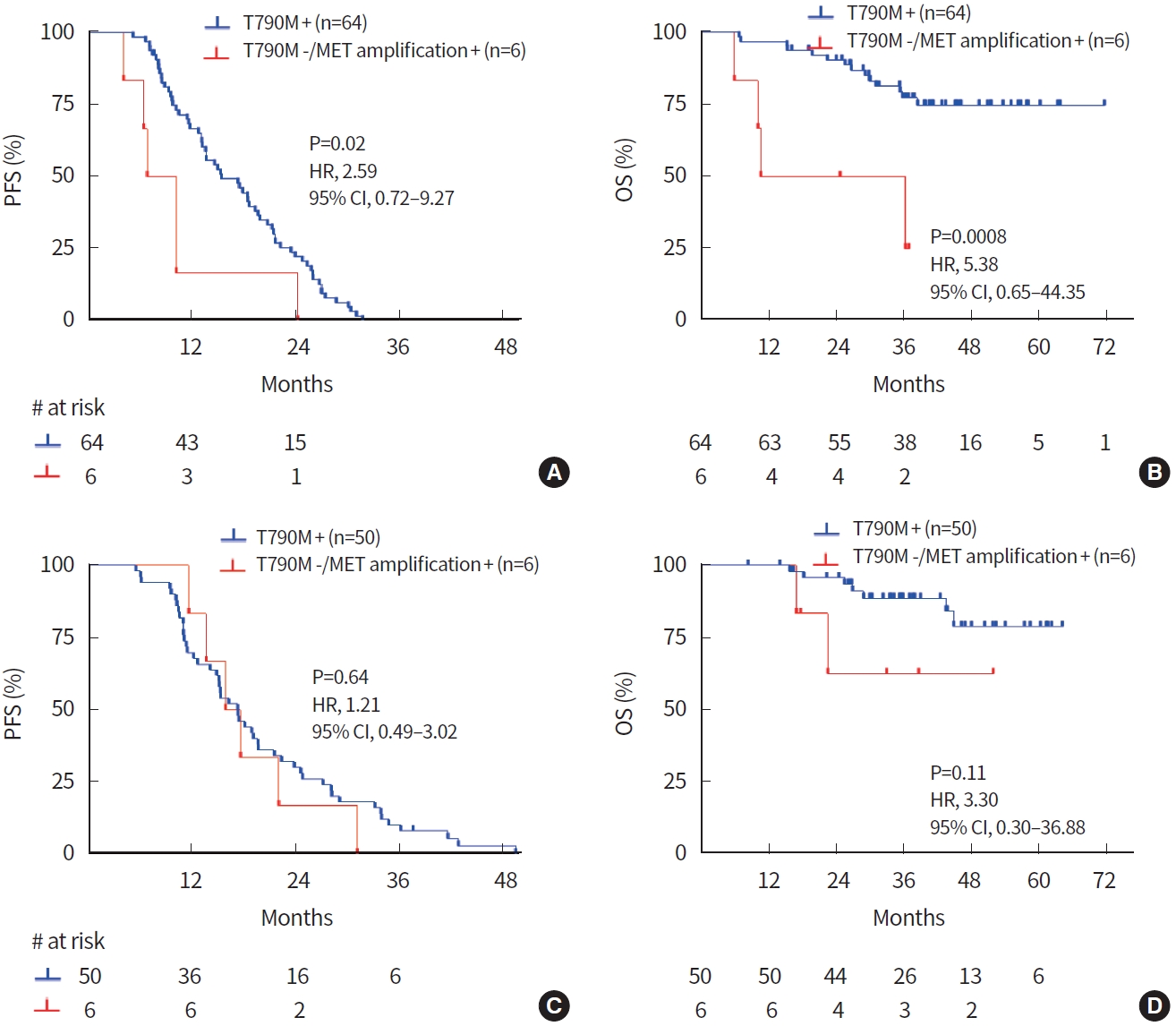
Prognostic significance according to mesenchymal-epithelial transition tyrosine kinase receptor (MET) amplification and epidermal growth factor receptor (EGFR) T790M mutation. In the 1st-generation EGFR tyrosine kinase inhibitor (TKI) group, T790M-positive patients showed significantly longer (A) progression-free survival (PFS) and (B) overall survival (OS) than did T790M-negative/MET amplification-positive patients. In the 2nd-generation EGFR TKI group, (C) PFS and (D) OS curves showed no significant difference according to T790M and MET amplification. HR, hazard ratio; CI, confidence interval.
DISCUSSION
In this study, we evaluated the MET status of 241 lung adenocarcinoma patients who received EGFR TKI therapy using biopsy samples at baseline and progression. Among them, 111 patients had paired biopsy samples and were separately analyzed to compare MET status change before and after treatment. To the best of our knowledge, this is the first study to compare the results of IHC and FISH for MET status in paired biopsy samples of EGFR TKI-treated patients.
In our population, 16.4% and 1.6% of patients showed MET overexpression by IHC and MET amplification by FISH before EGFR TKI treatment. However, these proportions increased after EGFR TKI treatment to 22.5% and 7.8%, respectively. These proportions are consistent with MET amplification prevalence approximated in previous studies. Paired biopsy samples showed similar results, suggesting that both IHC and FISH can be used to identify MET status alteration. However, MET overexpression by IHC might not always be reflective of MET amplification [17]. In acquired resistance mechanisms of EGFR TKI, T790M, and MET amplification have been found almost mutually exclusively [18]. When stratifying a study population according to presence of T790M mutation, T790M-positive patients had much lower MET overexpression and amplification by both IHC and FISH than did T790M-negative patients. Based on the well-known inverse relationship of T790M mutation and MET amplification, MET IHC showed high false positive prediction, and FISH showed more accurate detection for MET amplification. When MET FISH was performed to confirm the MET IHC result, some samples with strong intensity of IHC (3+) had low GCN (< 5) by FISH. In addition, 19 paired samples with MET IHC-positive (1–3+) at baseline showed MET IHC negativity at progression. However, there was no case of baseline-positive and progression-negative FISH-tested sample. Consistent with a previous report [17], these results suggest that MET amplification measured by FISH more accurately reflects development of MET amplification after EGFR TKI treatment than does IHC. In addition to FISH and IHC to test MET amplification, NGS allows identification of MET copy number with comprehensive high-throughput analysis of other alterations. However, copy number analysis based on NGS requires higher quantity tissue sample than FISH or IHC and has some disadvantages such as limited target region coverage, low sequencing efficiencies, and high cost. In addition, there are no clinically defined cutoff values for MET amplification using NGS. Thus, considering lack of consensus on the definition, FISH with recommended evaluation criteria might be the most suitable surveillance tool for predicting MET amplification.
MET amplification seems to be related to EGFR TKI resistance and worse overall prognosis, and its negative prognostic value has been confirmed in other studies and meta-analysis [16,19-21]. In survival analysis of patients treated with 1st-generation EGFR TKI, MET amplification by FISH showed predictive efficacy for survival benefits. However, patients treated with 2nd-generation EGFR TKI did not show any difference in PFS or OS according to presence of MET amplification. Considering co-occurring mutations such as MET exon 14 skipping and KRAS mutations in MET-amplified cohort [22,23], comprehensive molecular profiling using NGS will be need to explain the difference of survival benefit between 1st-and 2nd-genration EGFR TKI-treated patients. Comparing clinical outcomes of patients with either MET amplification by FISH or T790M mutation, patients with T790M had significantly longer PFS and OS than did patients with MET amplification. The superior outcome of T790M-positive patients might be due to use of 3rd-generation EGFR TKIs, which target T790M. Thus, this prognostic tendency might change if an effective MET inhibitor becomes widely available. This tendency was also identified only in patients treated with 1st-generation EGFR TKI not in those treated with 2nd-generation EGFR TKI. Considering previous in vitro studies showing that 2nd-generation EGFR TKI decreased EGFR and phosphoinositide 3-kinase (PI3K)/Akt activities and proliferation of T790M-positive cell lines [24], such drugs might have additional effects of blunting the negative prognostic effects of MET amplification. In a recent study [25], patient stratification by MET amplification had effect on PFS only with a 1st-/2nd-generation EGFR TKI but not with a 3rd-generation drug. Further studies will be needed to identify exact reasons for these discrepancies between EGFR TKI used or line of therapy.
MET TKI combinations with EGFR TKI demonstrated clinical activity against previously EGFR TKI-treated patients [26]. The combination of capmatinib with gefitinib demonstrated favorable overall response rate (ORR) (27%) in a phase Ib/II trial in patients with MET amplification pretreated with EGFR TKI [27]. In another phase Ib/II trial, the combination of tepotinib with gefitinib also showed longer PFS and OS than did standard chemotherapy in MET-amplified patients [28]. In the phase 3 randomized, double-blind, palcebo-controlled study of ARQ 197 plus erlotinib versus placebo plus erlotinib (MARQUEE) study, erlotinib plus tivantinib improved PFS over erlotinib monotherapy in previously treated EGFR-mutant NSCLC [29]. In contrast, an upfront combination of erlotinib and an anti-MET monoclonal antibody (emibetuzumab) failed to demonstrate a survival advantage compared with EGFR TKI alone in unselected patients with EGFR-mutant advanced NSCLC [30]. Several other clinical trials using concurrent EGFR TKIs and MET TKIs are currently under investigation in patients with EGFR-mutant NSCLC with MET amplification. These results suggest that identification of MET amplification-driven EGFR TKI resistance is important to better select patients for treatment.
Although this study provided the largest MET analysis using paired biopsy in patients with NSCLC who developed resistance after EGFR TKI treatment, there are several limitations. First, as this study was conducted retrospectively in a single center, there were potential biases. Second, though differences of clinical outcome were shown between patients treated with 1st- and 2nd-generation EGFR TKI, it was difficult to explain how MET affects this result. Therefore, further studies are needed to explain and validate these results.
In conclusion, our results confirmed MET amplification as an EGFR TKI-resistant mechanism in paired biopsy samples. Considering the false positivity of IHC, FISH should be recommended for accurate estimation of MET amplification.
Supplementary materials
Representative image of mesenchymal-epithelial transition tyrosine kinase receptor (MET) immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH). (A) MET IHC. The staining intensity was graded as negative (0, upper left), weak (1+, upper right), moderate (2+, lower left), and strong (3+, lower right). Scale bar: 100 μm. (B) Representative negative (left panel) and positive (right panel) case of MET FISH. Pictures taken at 20× (for IHC) and 60× (for FISH) magnification power.
Prognostic significance of mesenchymal-epithelial transition tyrosine kinase receptor (MET) overexpression (OE) by immunohistochemistry (IHC) in patients treated with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). In the 1st-generation EGFR TKI group, (A) progression-free survival (PFS) and (B) overall survival (OS) curves according to MET OE had no significant difference. In the 2nd-generation EGFR TKI group, patient stratification by MET IHC had no effects on (C) PFS and (D) OS. HR, hazard ratio; CI, confidence interval.
Acknowledgements
This research was supported by the National Research Foundation (NRF) grants (NRF-2021R1I1A1A01044209) and Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government(MSIT) (No.2021-0-02068, Artificial Intelligence Innovation Hub).
Notes
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conception or design: BMK, SP, MJA.
Acquisition, analysis, or interpretation of data: BMK, SP, SP, HAJ, JMS, SHL, JSA, YLC, MJA.
Drafting the work or revising: BMK, SP, MJA.
Final approval of the manuscript: BMK, SP, SP, HAJ, JMS, SHL, JSA, YLC, MJA.