Tau positron emission tomography in tauopathies: A narrative review
Article information

Abstract
Aggregation of misfolded tau in the brain is a major pathological feature common in various neurodegenerative disorders known as tauopathies, including Alzheimer’s disease, progressive supranuclear palsy, corticobasal syndrome, and dementia with Lewy bodies. Tauopathies are collection of diseases with varied overlapping symptoms and complicated manifestations. Consequently, it is crucial to be able to assess tau deposits in vivo. Over the past decade, tau-specific radioligands for positron emission tomography (PET) have been developed and tested, including first-generation compounds (e.g., 18F-THK5317, 18F-THK5351, 18F-AV1451, and 11C-PBB3) and second-generation compounds (18F-MK-6240, 18F-RO-948, and 18F-PI-2620). With the recent advances of tau PET tracers, assessing the pattern of tau deposition in vivo is possible. These methods will allow accurate diagnosis of tauopathies and monitoring of disease progression. In this mini review, we summarize current findings from studies using tau PET tracers regarding neuropathological characteristics, clinical implications, and potential applications of tau PET. We also discuss methodological considerations for appropriate use of these technologies and discuss what has been learned from these findings.
INTRODUCTION
Tau is a phosphoprotein encoded by the microtubule-associated protein tau (MAPT) gene on chromosome 17q21.3 and comprises 16 exons [1]. Alternative mRNA splicing of the MAPT gene leads to expression of six tau isoforms with three (3R) or four (4R) microtubule-binding repeats. Exclusion or inclusion of exon 10 results in either 3R or 4R, respectively [2-4]. Tau is involved in the formation and stabilization of microtubules in the nerve system, which is essential for neuronal stability and functioning. Phosphorylation of tau impacts its functionality [5]; however, hyperphosphorylation of tau causes and also increases the aggregation of tau into straight filaments, twisted ribbons, or paired helical filaments (PHFs) and, subsequently, into neurofibrillary tangles (NFTs), pick bodies, glial tangles, coiled bodies, and astrocytic plaques etc. An accumulation of abnormal tau in the human brain is the main pathological hallmark of neurodegenerative disorders that are now collectively termed “tauopathies” [6,7]. Many of the most common neurodegenerative diseases are classified as tauopathies, such as Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease, and chronic traumatic encephalopathy (CTE) [8,9], and they exhibit distinct regional distributions of abnormal tau deposition [10]. One of the main histopathological features in AD is aggregation of hyperphosphorylated tau into NFTs, along with the formation of amyloid-β (Aβ) plaques [11]. The regional distribution of NFTs follows a stereotypical pattern defined as “Braak stages”: stages I–II (transentorhinal), III–IV (limbic), and V–VI (isocortical association areas) [12]. In CBD and PSP, the distinct lobar and basal ganglionic tau deposits are different from those in AD [13,14]. In addition to the distinct regional distribution of deposition, tau isoforms are an important factor of tauopathy. Tau isoforms containing either 3R or 4R are present in approximately equal distributions in the normal brain and in patients with AD, tangle predominant senile dementia, and CTE [15,16]. The ratio is substantially altered in other tauopathies: 3R isoforms are dominant in Pick’s disease and 4R isoforms are dominant in CBD, PSP, and argyrophilic grain disease [16].
Despite advances in the research of tauopathies, many questions regarding tau deposition in the pathophysiology of these neurodegenerative diseases remains unclear. These diseases have substantial biochemical and pathological similarities and show considerable overlapping clinical phenotypes [8]. Therefore, accurate and early differential diagnosis of these diseases is a clinical challenge. Because of the invention of tau tracers for positron emission tomography (PET) [17], it is possible to visualize and quantify tau pathology in the living human brain. This approach has great potential in early diagnosis, especially in cases with ambiguous clinical symptoms. In addition, tau PET imaging can be useful for subject selection and as a surrogate outcome measure in clinical trials. Since the introduction of tau PET tracers, studies using tau PET have shown an exponential increase in the field of neurodegenerative diseases (Fig. 1). Despite the potential value of tau PET imaging, however, application of this tool in clinical practice is challenging. Its value in investigating pathological tau accumulation and in diagnosing tauopathies remains to be clarified, and the development of novel tau ligands is ongoing [18]. In this brief review, we summarize and discuss findings from recent studies on various tau PET ligands and future directions of tau PET imaging.
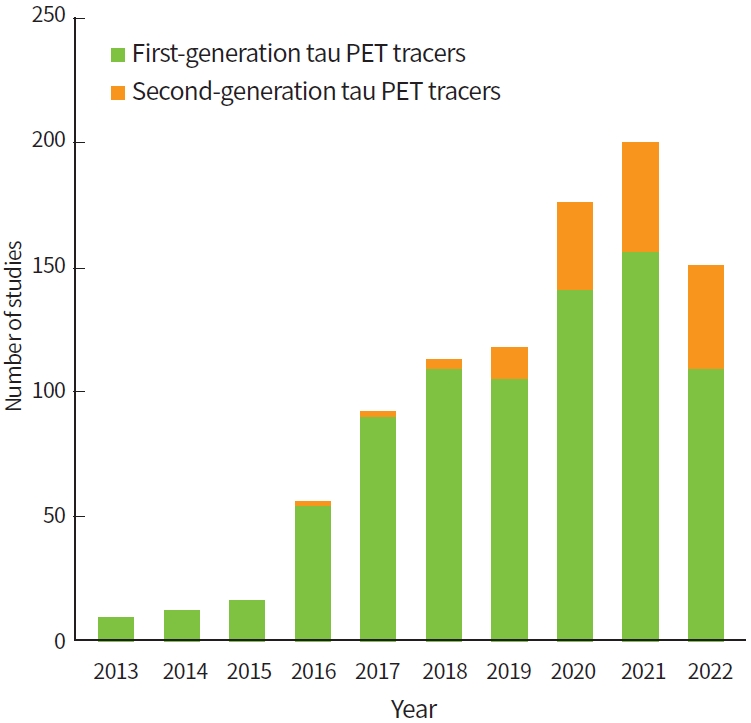
DEVELOPMENT AND CHALLENGES FOR TAU PET IMAGING
Development of PET tracers that specifically target the tau protein in the human brain presents a great challenge [19]. In addition to the general characteristics required for an ideal brain PET tracer [20-22], the requirements for tau PET tracer are even more strict.
Low toxicity, sufficient blood-brain barrier penetration, low non-specific binding, rapid uptake and clearance from the brain, and no radiolabeled metabolites in the brain are required for brain PET tracers [23]. Furthermore, the labelling of 18F instead of 11C is preferred due to the longer half-life [23-25]. In addition, tau PET tracers have additional challenges [26]. First, tau is largely an intracellular protein and must be able to cross the cell membrane, which limits its molecular size and lipophilicity [23]. Second, tau deposits are co-localized with Aβ plaques in AD. Therefore, tracers must be selective for tau deposits over Aβ [23,24]. For example, 2-(1-{6-[(2-[fluorine-18]fluoroethyl)(methyl)amino]-2-naphthyl}-ethylidene)malononitrile (18F-FDDNP) was a promising candidate tau tracer that was originally developed for AD. However, it failed due to a lack of selectivity as it binds to both Aβ and tau [27]. The problem is that current tau PET tracers share β-sheet binding properties, but Aβ has similar structural motifs to β-sheets. In addition, Aβ is co-localized with tau deposits at much higher concentration. Tracers may bind to Aβ with different affinities [23]. Third, tau deposits in various tauopathies contain different isoform compositions: 3R and 4R tau are present in AD, tangle predominant senile dementia, and CTE; 3R tau is dominant in Pick’s disease; and 4R tau is dominant in CBD, PSP, and argyrophilic grain disease [16]. These differences hinder the selectivity of tau PET tracers in differentiating tauopathies. Due to structural similarities, the development of PET tracers that bind exclusively to specific isoforms remains challenging.
For these reasons, while many molecules have been recommended as potential tau PET tracers over the past decade, the development and clinical implementation of tau tracers have been slow mainly due to lack of sufficient specificity and selectivity [28,29].
FIRST-GENERATION TAU PET TRACERS
A variety of molecules has been suggested and tested for tau imaging. Several tracers are now available in different stages of clinical development, which can roughly be categorized into first-generation and second-generation tau PET tracers.
First-generation tau PET tracers can be divided into three families according to chemical structure [23,30,31]: pyrido-indole derivative ligands (18F-AV680 and 18F-AV1451) [32,33]; aryquinoline derivative ligands (THK family: 18F-THK523, 18F-THK5105, 18F-THK5117, 18F-THK5351) [34-37]; and phenyl/pyridinyl-butadienyl-benzothiazole/benzothiazolium derivative ligands (11C-PBB3) [38-40].
18F-AV680 (formerly 18F-T808) and 18F-AV1451 (formerly 18F-T807, 18F-flortaucipir, and TauvidTM [Eli Lilly and Co.]) were developed by Siemens and are now owned by Eli Lilly. 18F-AV1451 showed better properties as a tau imaging agent [17,41] and became the most widely applied tracer. It was recently approved by the U.S. Food and Drug Administration (FDA) for clinical use in the United States as TauvidTM (Eli Lilly and Co.) [42]. To date, 18F-AV1451 is the first and only FDA-approved tau PET imaging agent. The THK family (18F-THK523, 18F-THK5105, 18F-THK5117, 18F-THK5351) was developed at Tohoku University in Japan [25,34,36,37,43]. Among the tracers, 18F-THK5351, the newest derivative in this family, exhibits better characteristics including a higher signal-to-noise ratio and lower white matter (WM) retention. However, the first three of these tracers have been precluded from the next stages of clinical development due to high retention in WM [44]. 11C-PBB3 (Chiba, Japan), which is derived from the same tracer family as the Aβ ligand Pittsburgh Compound B (PIB), has been used to image AD and non-AD tauopathies [38,39]. However, it exhibited low specific binding and radio-labelled metabolites capable of crossing the blood-brain barrier [45].
Despite the initial success of first-generation tau tracers, they exhibit some drawbacks, including low specific binding, off-target binding, and radiolabeled metabolites entering the brain. Off-target binding is a critical flaw as a specific tau PET tracer (see section of ‘off-target binding’). Given that 18F-AV1451 is a currently established and approved tau PET tracer, we focus on this tracer.
Neuropathologic correlation and characteristics
It is crucial to verify that the PET tracer signal detected in vivo corresponds to pathology. For this purpose, autoradiography studies on postmortem brain tissue have been performed to assess the role of tau PET tracer signal. Most first-generation tracers bind to the tau aggregates formed in AD (AD tauopathy, a mix of 3R/4R tau isoforms) [46-48]. However, they exhibit lower affinity for the 3R or 4R isoforms that characterize many non-AD tauopathies and may be related to the lower tau aggregate densities that hinder detection using tau PET [4].
Supporting 18F-AV1451 as the most established tau PET tracer, extensive studies have validated neuropathologic correlation of tau PET [49-53]. A large end-of-life study [49] and extended case series [50,51] demonstrated that 18F-AV1451 more accurately detects AD tauopathy in individuals in more advanced Braak stages (i.e., Braak >IV; the accuracy for detecting tau deposit corresponding to Braak stages V and VI was 87.5% [95% confidence interval, 77.2% to 93.5%] [49]). There is a strong correlation (R2 range, 0.66 to 0.76) between tau PET level and quantitative neuropathologic tau burden in corresponding brain regions [52,53]. In addition, in MAPT mutation carriers with mixed 3R/4R tau pathology, similar to AD, there is a strong correlation (R2=0.86) between the tau PET signal and the postmortem neuropathologic tau burden [54,55].
Unlike AD tauopathy, the evidence supporting the in vivo correspondence of PET signal to tau aggregation in non-AD tauopathies is less clear. Studies on non-AD tauopathies have shown mixed results. For 18F-AV1451, while studies have reported differences of detected in vivo PET signal between controls and individuals clinically diagnosed with 4R tauopathies (PSP, CBD) are part of non-AD tauopathies [55-60], binding to non-AD tauopathies has been limited to postmortem studies [4,50,61].
Studies showed that 18F-AV1451 binds to motor-related subcortical gray and WM structures in patients with corticobasal syndrome, which distinguishes patients from controls [55-57]. However, only a few autopsy-confirmed cases were reported with either moderate-to-strong correlation (R2 range, 0.59 to 0.79) [62,63] or a limited correlation of PET signal with tau pathology [50].
There are significant differences of tracer retention between controls and clinically diagnosed PSP patients in studies using 18F-AV1451 PET scan [55,58-60]. Tracer retention is predominant in the substantia nigra and basal ganglia, which also show off-target binding to 18F-AV1451, complicating interpretation [4]. There was no significant correlation between cortical 18F-AV1451 PET signal and neuropathologic 4R tau [61], and little binding outside the off-target regions was observed [50]. The number of autopsy-confirmed cases was too small to draw any conclusions [50,61,64].
In summary, the currently established first-generation PET tracer binds to AD tauopathy in the more advanced Braak stages (>IV). However, the diagnostic value of the tracer for AD tauopathy may be suboptimal. NFTs may be present at levels suitable for a neuropathological diagnosis of AD, Braak staging II tau pathology in the presence of at least moderate levels of cortical amyloid pathology, even in patients with a negative 18F-AV1451 scan. Besides, the potential of the tracer in quantifying and visualizing non-AD tauopathies in vivo is highly limited since tracer binding in most non-AD tauopathies is weak and overlaps to a large extent with known off-target binding regions [4].
Quantification of tau retention in the brain is of great interest in the clinical applicability of tau PET imaging. Therefore, determination of an optimal method for quantifying tau deposit is essential [45,65-69]. In vivo kinetic models using arterial sampling are the “gold standard” for accurate assessment of the pharmacokinetic properties of tau PET tracers. Kinetic modeling with arterial sampling in humans has been performed for first-generation tracers [45,67,68,70,71], except 18F-THK5351. However, finding and optimizing a less invasive and less laborious method that can be easily implemented in the clinic is of great value. In this regard, several studies on less invasive quantification methods, using reference tissue models without arterial sampling, have been performed. Studies have also examined the optimal time interval for quantification and validated semi-quantitative quantification approaches such as standardized uptake value ratio (SUVR) [3]. All tracers showed the feasibility of using reference tissue models and SUVR values as a reliable measurement of tau deposit in vivo. A previous study has reported good correlations between SUVR and plasma-input kinetic model-derived parameters [71], which has a great value for future applicability, since these methods are more suited for use in clinical settings. Cerebellar regions were selected as the reference tissue in all such models because they are relatively spared from tau deposits in AD until late in the disease course [3,12].
Off-target binding
First-generation PET tracers show off-target binding to such an extent that it impedes the specificity of these tracers to detect tau pathology. Sources of off-target binding vary widely and include monoamine oxidase (MAO), amyloid deposit, calcifications, iron, and microhemorrhages [31,72].
11C-PBB3 and the 18F-THK ligands show substantial off-target binding to amyloid deposits and MAO-B, which limits the use of these traces for selective tau pathology detection [31,73,74]. 18F-AV1451 shows off-target binding at the substantia nigra, eye, basal ganglia, longitudinal sinuses, pituitary, and choroid plexus (Fig. 2) [47,75,76]. The most apparent targets are neuromelanin in the substantia nigra and retinal pigment epithelium in the eye [47]. MAO is also a potential source of off-targeting, as shown in in vitro study of 3H-AV1451 binding to MAO-A and MAO-B [77]. However, in vivo studies using 18F-AV1451, MAO does not appear to be a significant binding target [78,79]. Interestingly, 18F-AV1451 also shows elevated tau PET signal in patients with clinical syndromes typically associated with TAR DNA-binding protein 43 (TDP-43) type C rather than tau pathology, such as the semantic variant of primary progressive aphasia [80,81].

Off-target binding of first-generation tau positron emission tomography (PET) tracers. (A) 18F-AV1451 PET images in health volunteer showing off-target binding in basal ganglia, choroid plexus, retinal, and pituitary. (B) 18F-THK5351 PET images showing high signal intensity in basal ganglia, which is explained by binding to monoamine oxidase B.
SECOND-GENERATION TAU PET TRACERS
As a result of continued efforts to minimize the off-target binding observed in first-generation tau PET tracers, second-generation tracers, including 18F-MK6240, 18F-RO948 (18F-RO6958948), 18F-PI2620, 18F-genentech tau probe 1 (GTP1), and 18F-JNJ-64326067 [82-87], are now available. Many potential tracers are being developed and analyzed for use as biomarkers. In this review, we focus on several tracers that have passed the initial validation stage for clinical use.
18F-MK6240 is a pyridine isoquinoline amine derivative that was developed by Merck. This tracer shows good characteristics for in vivo imaging, including an appropriate dynamic SUVR range, desirable kinetics, significant binding in regions known to contain AD tauopathy, and correlation with clinical endpoints [88].
18F-RO948 and 18F-PI2620 are derivatives of 18F-AV1451, with a pyrido-indole-based structure. They can distinguish AD subjects from healthy controls and have promising characteristics as tau PET tracers [18,42]. 18F-RO948, developed by Roche [18,42], exhibits significantly higher tracer retention in patients with AD compared with controls, lack of radiolabeled metabolites entering the brain, and no defluorination [42,89]. 18F-PI2620, developed by Piramal Imaging, has robust uptake and fast wash-out in AD subjects and focal asymmetric uptake in AD tau-bearing areas. Importantly, preclinical characterization of 18F-PI2620 has shown strong binding in Pick’s (3R) and PSP (4R) non-AD tauopathies [90].
18F-GTP1, developed by Genentech, is under evaluation in a longitudinal natural history study. Preliminary cross-sectional results show an association between 18F-GTP1 uptake and cognitive deficits in AD but also notable off-target binding in the basal ganglia [91].
Neuropathologic correlation and characteristics
While first-generation tracers have shown lower affinity for the 3R or 4R isoforms of tau that characterize non-AD tau pathology [18,46-48,86], for some of the second-generation tracers, there is evidence of binding to the 3R or 4R isoforms from autoradiography studies [92,93]. This neuropathological correlation could facilitate more reliable diagnosis of non-AD tauopathies by tau PET imaging. In addition, these tracers show promising binding profiles with lower off-target binding in the target regions for PSP or multiple system atrophy compared with first-generation tracers [75,76]. 18F-PI2620 has the capability of binding to the 4R isoform in tissue of individuals with PSP [92]. A multicenter study reported significantly increased 18F-PI2620 binding in predefined PSP target regions in clinically diagnosed PSP patients, indicating the value of this tracer in differentiating suspected patients with PSP [92]. However, despite some evidence of binding to 4R tau pathology on autoradiography [92], a recent study showed limited binding of 18F-PI2620 to 4R tau pathology [94]. Notably, the in vivo second-generation tau PET signal only partially reflects 3R or 4R tau pathology. More data are needed to validate each of these tracers as an in vivo imaging biomarker of tauopathy.
Off-target binding
The off-target binding profiles of second-generation tracers still vary but to a lesser degree and extent compared with first-generation tracers. The most apparent off-target binding target of 18F-MK6240 is neuromelanin in the substantia nigra and retinal pigment epithelium [86]. In addition, as a derivative of 18F-AV1451, 18F-RO948 is associated with binding to neuromelanin, like its predecessor [4,18]. In head-to-head studies against 18F-AV1451, 18F-MK6240 and 18F-RO948 show reduced binding to the basal ganglia, longitudinal sinuses, pituitary, and choroid plexus [4,75,76]. In contrast, they show substantial off-target binding to the meninges and skull, especially in women [4,95,96]. Off-target binding profiles of 18F-PI2620 show involvement in the meninges, skull, and venous sinuses [94]. Details of tracers are summarized in Table 1 [18,23,33,43,45-47,77,82,89-91,95,97-106] and Fig. 3.

Binding characteristics of representative first- and second-generation tau tracers
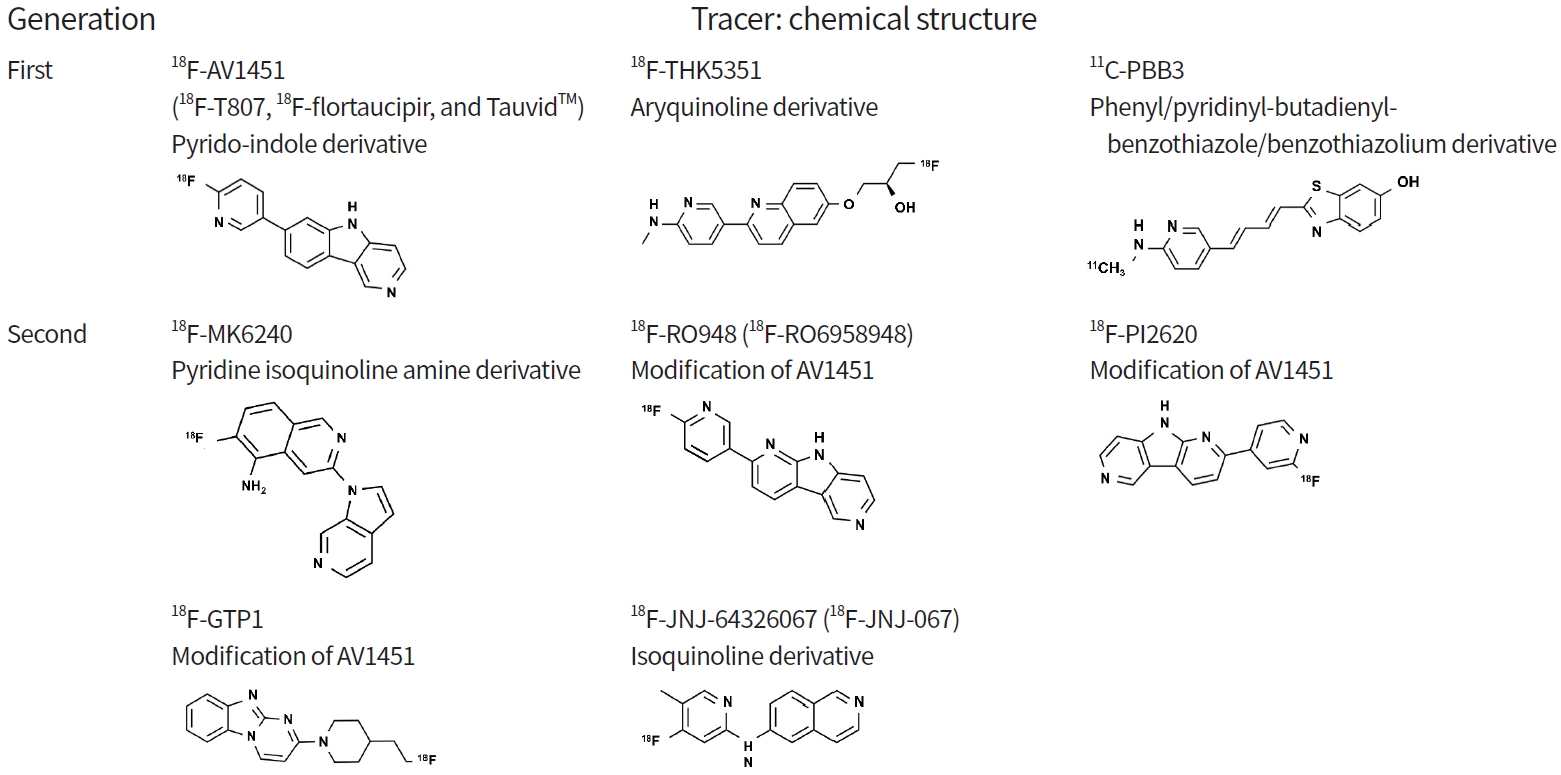
Chemical structure of representative first- and second-generation tau tracers.
CLINICAL IMPLICATIONS
Implications in AD
Because of considerable overlap of clinicopathological features across neurodegenerative disorders related with tauopathy, diagnosis and differentiation of the diseases are challenging. In secondary tauopathies such as AD, tau pathology is considered a response to other pathological events like Aβ [107,108]. In primary tauopathies such as PSP and CBD, abnormal aggregation of tau is the major hallmark of the pathology. With the emergence of tau PET imaging, diagnosis of tauopathies has received increasing attention. Given that most neurodegenerative dementias, including AD, are characterized by tauopathy, tau PET imaging may be able to detect many dementias in a diagnostic setting [109]. Furthermore, tau PET tracers, such as 18F-AV-1451, 18F-MK6240, 11C-PBB3, and 18F-RO948, showed substantial diagnostic performance for distinguishing AD dementia from non-AD neurodegenerative disorders, with high sensitivity and specificity [40,110-112]. However, although tau PET imaging has the potential to discriminate AD dementia patients from cognitively normal (CN) individuals, the individual diagnostic value of tau PET in the clinic has not been fully assessed. In a recent study, tau PET showed a similar impact on diagnostic confidence to amyloid PET. However, negative tau PET showed a lesser impact on etiological diagnosis compared with negative amyloid PET [113].
Of note, there is a strong relationship between the symptomatology of AD and the pattern of tau PET deposition. Retention of tau PET tracer is prominent in the clinically affected regions of clinical variants of sporadic AD, such as posterior cortical atrophy, logopenic variant of primary progressive aphasia, or behavioral/dysexecutive variant [114-116]. Therefore, tau PET imaging can be useful in accurately detecting atypical variants of AD, which show highly distinct patterns compared with typical AD [114,117]. There is also a correspondence between retention of tau PET tracer and autosomal dominant AD, a subgroup of AD that is estimated to account for approximately 1% of AD cases [118]. Tau deposits were associated with symptom onset, and the regional binding pattern was different from that of non-autosomal dominant AD [119]. Given its high specificity, tau PET may be useful to discriminate stages of AD, including preclinical, prodromal, mild cognitive impairment (MCI), and dementia (Fig. 4) [3,120]. While amyloid PET is likely not sufficient to discriminate between symptomatic stages of AD, the intensity and spatial pattern of tau PET tracer retention have the potential for such discrimination [3,121,122]. However, previous studies have demonstrated substantially decreased discriminative accuracy of tau PET at the prodromal stage of AD compared with more advanced stages [101,123,124]. Therefore, more data are needed to draw conclusions.

Patterns of uptake observed with 18F-AV-1451 at different disease stages. Axial positron emission tomography (PET) images of 18F- AV1451 from one cognitively normal with Korean-Mini Mental State Examination (K-MMSE) of 28 (A), two mild cognitive impairment with K-MMSE of 22, 21 (B, C, respectively), and one Alzheimer dementia with K-MMSE of five (D) patients. Signal intensity and extent increase with disease severity, which are successively involving temporal, parietal, frontal, and occipital cortices.
Although the presence of Aβ deposit is considered necessary for a diagnosis of AD, such deposits are prevalent among the elderly [125]. Furthermore, individuals with amyloid positivity can remain in a cognitively unimpaired state for decades before they experience cognitive decline [125,126]. Amyloid PET seems to be a suboptimal predictor of clinical progression in cognitively unimpaired individuals. However, tau PET has been found to have great value in predicting preclinical and prodromal cognitive changes [127]. Interestingly, in a longitudinal study with 129 cognitively unimpaired participants who underwent amyloid and tau PET scans and were followed for a minimum of 2 years, the amyloid-positive and tau-positive subjects showed cognitive decline over the course of follow-up, whereas both amyloid-negative/tau-negative subjects and amyloid-positive/tau-negative subjects remained cognitively unimpaired. Only one subject classified as amyloid-negative and tau-positive was cognitively unstable. Both tau and amyloid showed an association with cognitive decline; however, only tau remained significant when both variables were included in the analysis. This result supports the notion that cognitive decline is predominantly driven by tau pathology and consistent with observations that tau deposition in the neocortex manifests as the disease progresses [127,128]. Tau PET is superior to amyloid PET in predicting preclinical and prodromal cognitive changes [127].
Tau PET has great potential as a prognostic marker in symptomatic stages of AD. The intensity of amyloid and tau PET level at baseline scan are strongly associated with more rapid cognitive decline [129,130]. Positivity on 18F-AV1451 PET scan was a strong indicator of future cognitive decline in patients with MCI and AD [131]. Notably, the specific distribution of baseline tau PET deposit was predictive for future brain atrophy even at the single-patient level among those with MCI and AD and was particularly strong in younger patients [132].
In summary, tau PET may be useful to detect atypical variants and to discriminate stages of AD. The retention of tau PET tracer is a prognostic marker in the prodromal stage as well as in the symptomatic stage. Tau PET positivity, especially in combination with amyloid PET positivity, is an excellent biomarker in predicting short-term progression of cognitive decline in individuals with risk of AD, which supports the notion that tau pathology is a key driver of cognitive decline [127,128,132]. Tau PET may be potentially used for individualized risk profiling and state assessment in clinical practice.
Implications in non-AD tauopathies
Neurodegenerative disorders that are primary tauopathies have distinct patterns of spatial distribution of tau deposits, allowing differentiation of tauopathies by tau PET imaging. Imaging of the regional distribution of tau pathology may be useful in distinguishing diseases classified as non-AD tauopathy such as CBD, PSP, and primary age-related tauopathy (PART).
Tau PET may be helpful in diagnosing CBD and PSP, which are types of atypical parkinsonism and have substantial clinicopathological overlap. These diseases are characterized by deposition of abnormally hyperphosphorylated tau predominantly in subcortical regions, in contrast to AD [13,14]. Concordance between retention of tau tracer in PET imaging and pathological patterns of tau deposition was reported in patients with PSP and CBD. In patients with a clinical diagnosis of PSP, high retention of tau PET tracers was reported in the basal ganglia, thalamus, midbrain, and cerebellum; this is consistent with the areas expected based on previous neuropathological studies [58-60,133,134]. In Aβ-negative patients with clinical diagnoses in the CBD spectrum, tau PET tracers 11C-PBB3, 18F-THK5317, and 18F-THK5351 were predominantly located in WM and the basal ganglia [82,133,135]. 18F-RO948 PET also can visualize representative patterns across diagnosis groups including CBD, PSP, and AD [4]. Importantly, tau PET may be superior to other AD biomarkers, including magnetic resonance imaging (MRI) and most biofluid markers, in regard to early and reliable diagnosis [110,123,136,137].
Tau PET also has the potential utility for differential diagnosis between dementia with Lewy bodies (DLB) and AD. DLB is characterized by α-synuclein aggregates, known as Lewy bodies. However, tau deposits similar to those in AD pathology are also common, and clinical distinction between DLB and AD can be challenging [138-140]. 18F-AV 1451 retention in patients with DLB was higher than that in controls but varied widely [141]. In addition, the probable DLB patient group showed lesser 18F-AV 1451 retention than the probable AD group, and retention in medial temporal lobe was highly distinctive, which allows researchers to distinguish AD from probable DLB [142].
PART is a pathologic diagnosis that shows NFTs (Braak stage ≤4) with low, if any, amyloid burden [143] and is attracting attention with development of tau PET. PART was thought to represent the equivalent of the previously described “suspected non-Alzheimer’s pathophysiology” (SNAP), but this is debatable [144]. Investigations using tau PET supported the hypothesis that SNAP is the in vivo counterpart of PART. Studies have described cases possibly representative of PART, Aβ-negative cases in CN individuals with focally elevated cortical tau PET tracer retention [4,145-147]. However, another study reported contrasting findings. In that study, CN individuals with SNAP did not exhibit evidence of elevated tau, which suggests that this biomarker construct does not represent amyloid-independent tauopathy [148].
Together, these findings support the potential of tau PET tracers for differentiating non-AD tauopathies. However, some tau PET tracers might bind to non-target molecules and might not bind to substantially to tau pathology [46,47]. The tracers experience varying degrees of off-target binding at various locations including the substantia nigra and basal ganglia, which hinders the specificity of these tracers to detect tau pathology and can limit the diagnostic performance for tauopathies. In addition, the current diagnostic utility of tau PET in diagnosing individual patients is unclear. Thus, further investigation for each tracer is needed to assess the value of tau PET for non-AD tauopathies.
METHODOLOGIC CONSIDERATIONS FOR TAU PET IMAGING ANALYSIS
Region selection for determining tau PET positivity
Tau retention status in vivo imaging has been assessed using quantitative thresholds or visual assessment and usually is reported as binary classification [4,149-151]. The results are significantly influenced by the methodologies for classification of PET images [150] and require proper selection of brain regions in which positivity will be determined [151]. However, there is no established methodologic approach, which limits comparisons between studies. Given that the tau PET signal is substantially affected by factors including tau isoforms, off-target binding, and disease stage, the definition of tau PET positivity requires further study.
Disease stage is important and influences the region selection for classification, especially in AD. Regions of the temporal lobe cortex are candidates for positivity in the early stage of AD and the temporoparietal cortex is a candidate for positivity in later stages of the disease [4]. In cases with low accumulation of tau, the PET signal is not as easily distinguished from background noise. Consequently, the positivity of tau PET may be more affected by region selection in early stages of the disease. At the preclinical disease stage, the entorhinal or transentorhinal cortex has been proposed as a potential reference region (RR) to define tau PET positivity since it is typically the first region in which tau PET tracers can detect tau accumulation [4]. However, autopsy studies have reported that entorhinal tau deposit commonly occurs in older individuals without Aβ pathology, in a condition referred to as PART [143]. This indicates that tau pathology in the entorhinal cortex is not specific to AD, and the region is not appropriate for assessment in the early stage. A temporal meta-region of interest (ROI) consisting of the entorhinal, fusiform, and inferior and middle temporal cortices is another candidate and is more specific to AD [4]. However, this ROI may be less sensitive in early stages. In previous studies with cognitively unimpaired individuals with Aβ positivity, only approximately 5% to 10% of the individuals are classified as tau-positive [110, 152]. Interestingly, compared with temporal meta-ROI quantification, visual assessment shows better sensitivity in detecting tau positivity. Even at subthreshold levels of tau PET signal, visual positivity is possible [153]. In addition, although individuals with a visually positive signal might not meet the quantitative threshold for positivity, they tend to show positivity in other AD biomarkers including amyloid PET and cerebrospinal fluid (CSF) phosphorylated tau concentration [153].
The temporoparietal cortex has an advantage in that it can include a heterogeneous distribution of tau pathology from the typical presentation of Braak staging and atypical presentations such as posterior cortical atrophy and logopenic variant primary progressive aphasia [114,117].
Selection of the reference region for tau PET uptake measurement
SUVR is the most common semi-quantitative method to measure radiotracer uptake in an image and is calculated as degree of radiotracer uptake in a target ROI with respect to that in a RR [154,155]. SUVR is a convenient and useful tool for regional comparisons within a subject as well as between subjects; thus, SUVR has been widely used in studies based on brain PET images. As shown in the calculation formula, RR is the key factor that can significantly affect the overall SUVR measurement. Selection of an optimal RR is very important for accurate measurement with low variance [154]. Characteristics of an optimal RR are as follows. First, the region has to be devoid of target molecules that bind with radiotracer or are involved in the pathological process of disease. Second, the region should be free of off-target binding. Last, to minimize spill-in of radioactivity, the region needs to be distant from the region of tested radiotracer measurement.
Cerebellar gray matter satisfies the conditions required for tau PET studies, with absence of tau pathology in neuropathologic studies [53] and low variance in controls [72]. The mid-portion of the cerebellar gray matter such as the inferior cerebellar cortex or cerebellar crus is preferred as an RR since it can minimize spill-in from the occipital lobe and avoid off-target binding in the superior parts of the cerebellar vermis, observed with some other tracers [4,156]. Inferior cerebellar gray matter is a sensitive RR in discriminating group differences cross-sectionally. In a recent study with clinically unimpaired (CU) amyloid-negative individuals, CU amyloid-positive individuals, and MCI amyloid-positive individuals, this RR resulted in robust group differences, along with significant associations with CSF phosphorylated tau [157].
Interestingly, eroded WM may be more sensitive for longitudinal analyses. An eroded subcortical WM or eroded subcortical WM cerebellar composite RR may be more suitable for longitudinal analyses examining regional patterns of change [157-160]. The eroded WM RR more consistently detected significant change over time [157]. However, given that WM has a risk of spill-in due to its close proximity to the cortex, this RR needs to be verified in further studies with individuals with high cortical tau deposition [4].
An optimal processing approach that would minimize the influence of off-target signal is required. However, target or RR selection to reduce off-target signal should be performed carefully to avoid exacerbation of noise in SUVR data. A recent study with 18F-MK6240 reported that tau SUVR data are not obfuscated by noise, although off-target signal introduces noise. It is likely that off-target signal contaminates both reference and target regions, and the influence of off-target signal largely is cancelled out in SUVR data [161].
Partial volume correction
The partial volume effect (PVE) reduces the quantitative accuracy of an imaging system and is introduced by finite spatial resolution processes of imaging modalities. PET has poor spatial resolution compared with other imaging modalities such as MRI or computed tomography. The PVE is of concern and leads to overestimation or underestimation of radiotracer intensity estimates in PET imaging. Due to PVE, especially in small objects, acquired radioactivity from the object by the PET system differs from the ideal [162]. The PVE is a combination of two distinct phenomena: image blurring and image sampling [162]. Image blurring caused by finite spatial resolution of the imaging system results in spillover between regions and reduction of maximum activity in measured images. Typically, “spill out” of radioactivity into surrounding tissue from a high-activity region leads to underestimation of tracer uptake estimates, while “spill-in” into voxels of interest from a high-activity region leads to overestimation of radiotracer uptake estimates. Image sampling resulted from discrepancy of the actual contours of the radiotracer distribution and the radiotracer distribution is sampled on a voxel grid of PET imaging system. The actual contours of the object containing radiotracer do not match those of the voxels. Therefore, each voxel at the boundary of the target tissue represents the underlying tissues. In other words, the intensity of a particular voxel reflects the tracer concentration not only of the tissue within the voxel, but also that of the surrounding area.
In brain studies with PET, PVE is an important issue because it can influence SUVR measurement. For example, when the size of the target tissue region is small and the RR is adjacent to a high-radioactivity region, SUVR of the target region is likely to be measured as lower than actual. Therefore, partial volume correction (PVC) methods are needed for PVE. In a recent longitudinal cohort study with tau PET data, PVC improved the discriminative accuracy between cognitively impaired and unimpaired individuals cross-sectionally [158]. PVC also reduced the impact of choroid plexus off-target binding on hippocampal signals [148]. The geometric transfer matrix (GTM) method and more novel PVC methods modestly improved the diagnostic accuracy of tau tracer deposit in the hippocampus and the correlations between hippocampal SUVR and cognitive measures [163,164]. In addition, in a recent longitudinal cohort study with tau PET data, among five PVC methods (none, two-compartment, three-compartment, GTM, and tau-specific GTM variant), two-compartment voxel-based PVC showed the most favorable results [158]. However, consensus on the use of PVC has not been reached in tau PET studies. Although an obvious SUVR increase in tracer retention is measured with PVC data, whether PVC has a significant effect on the main results of studies (e.g., diagnostic performance, correlation with cognitive function) [4] and a standard PVC method have not been established. However, despite lack of consensus on the use of PVE in tau PET studies, it is increasingly recommended to report results both with without PVC.
CONCLUSION
Tau PET imaging enables visualization and quantification of tau pathology, indicating that tau PET imaging has great potential as a useful diagnostic tool for tauopathies. This technology provides great opportunity to elucidate previously unstudied paths of tau pathology in the living human brain and improves understanding of neurodegenerative disorders. For example, temporal and spatial information of tau burden could reveal a relationship with Aβ. These findings may be helpful to address how tau causes cognitive decline in AD, whether via synergistic interactions with Aβ or independently. A growing number of studies has provided valuable results on the usefulness of tau PET, including the ability to detect AD dementia, discriminate stages of AD, distinguish AD from other neurodegenerative diseases, predict the cognitive consequences of AD, and differentiate primary tauopathies from one another. Although the usefulness and diagnostic accuracy of tau PET at an individual level are not established, imaging has shown promise for detection of AD among cognitively unimpaired individuals, for prognostic use in AD, for distinguishing AD dementia from non-AD neurodegenerative disorders, and for differentiating non-AD tauopathies such as CBD and PSP from controls at a group level. However, there are several challenges that limit the utility of tau PET. First, off-target binding is a major drawback that weakens the possibility of tau PET as a diagnostic tool. Tracers can present large binding overlap with known off-target binding regions, which impedes reliable detection and quantification of tau burden especially in non-AD tauopathies. Selection of the optimal RR and application of PVC methods are also important methodological issues that could affect the potential use of tau PET imaging. Over the past decade, there have been continued developments for better tau tracers with improved sensitivity and specificity for tau pathology without off-target binding. These first- and second-generation tracers are now under investigation. More data are required to fully evaluate the potential of these tracers as imaging biomarkers in accurately characterizing tau burden. Additionally, it will be necessary to discuss further directions in the field of tau PET research. Head-to-head comparison with other biomarkers such as CSF and plasma biomarkers (e.g., p-tau and the Aβ42/40 ratio) is an important step to correctly interpret the results of different biomarkers and is required to develop an appropriate guide for use. Recent studies have shown that tau PET tracer binding correlates strongly with CSF tau biomarkers [165,166]. Establishing a standard method of tau PET measurement has great value for patient selection and treatment monitoring in clinical trials. Long-term study design with a more diverse population of patients could be helpful to further our understanding of the role of tau PET.
Notes
Sang Won Seo has been editorial board of Precision and Future Medicine since December 2017. He was not involved in the review process of this review aricle.
AUTHOR CONTRIBUTIONS
Conception or design: HL, YG, SWS, SHM.
Acquisition, analysis, or interpretation of data: HL, YG.
Drafting the work or revising: HL, SHM
Final approval of the manuscript: HL, YG, SWS, SHM.