Implication of hepatocyte dedifferentiation in pathogenesis and treatment of hepatocellular carcinoma
Article information
Abstract
Liver cancer is the fifth most common malignancy worldwide and the third most common cause of cancer-related death. Five hundred million individuals are infected by hepatitis B or C virus. Of these cases, some will progress to liver failure and cancer. Systemic treatments against liver cancer show poor efficacy mainly because of high resistance and poor response to therapeutics and high degree of tumor heterogeneity. Although progress has been made in combinational treatment of liver cancer, no effective disease-modifying therapies for hepatocellular carcinoma (HCC) have been developed to date. A comprehensive understanding of HCC pathogenesis will enable identification of biomarkers and therapeutic targets and provide insights into mechanism-based strategies for HCC treatment. However, the identification of cellular origins of liver cancer remains a challenging issue. This review focuses on HCC heterogeneity that escapes current treatment strategies and causes disease recurrence to provide a better understanding for the biology of liver cancer.
INTRODUCTION
Given the fundamental principle that cancer must arise from a cell that has the potential to divide, two major nonexclusive hypotheses have been proposed for the cellular origin of cancer development and malignancy: (1) it arises from stem and progenitor cells; and (2) it comes from dedifferentiation. The liver is subjected to routine exposure to injury and damaging agents via portal vein and cells [1]. It possesses a strong regenerative potential which contributes to reprogramming for tissue repair or intrahepatic tumor propagation. Whether hepatocellular carcinoma (HCC) originates from mature hepatocytes, stem and progenitor cells, or both remains controversial. Importantly, upon liver injury/damage, hepatocytes will undergo dedifferentiation and express genes that are characteristic of undifferentiated stem and progenitor cells. A widely-studied model supports that dedifferentiation of hepatocytes is the major point of origin of liver regeneration, although hepatic progenitor cells (HPCs) also play critical roles in liver development and repair [2-6]. Although the activation of HPCs (also called oval cells in rodents) also facilitates hepatic carcinogenesis [7,8], HCC mostly arises from dedifferentiation of fully-differentiated mature hepatocytes with ability to divide (Fig. 1) [9,10].
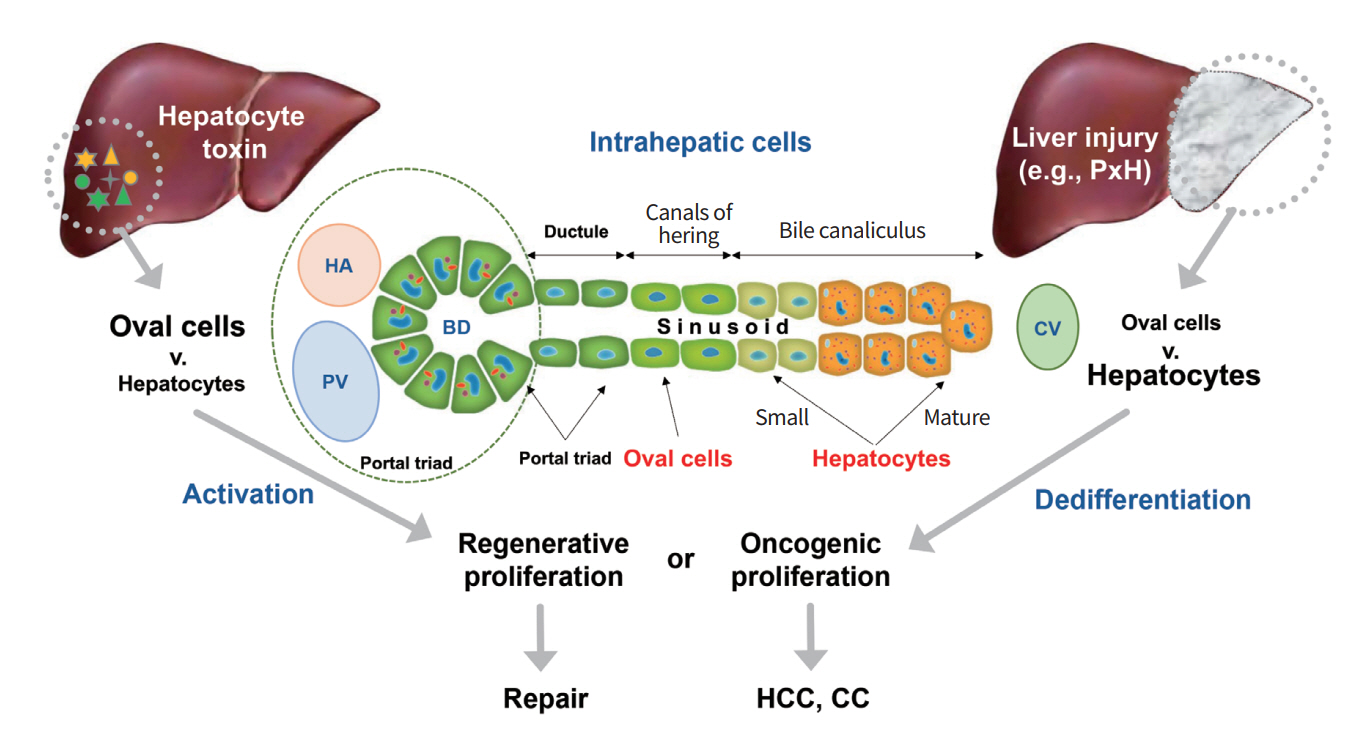
Model of liver regeneration by hepatic dedifferentiation and progenitor cell expansion following various types of liver injury/damage. PxH, partial hepatectomy; HA, hepatic artery; BD, bile duct; PV, portal vein; CV, central venule; HCC, hepatocellular carcinoma; CC, cholangiocarcinoma.
Most of the blood that enters the liver flows from the spleen, pancreas, and intestines via the portal vein. Thus, the liver is subjected to routine exposure to damaging agents in the bloodstream. Upon liver damage, hepatic cells will proliferate and rapidly regenerate large parts of the damaged organ in vivo. Different mechanisms of liver regeneration have been described using various injury models (Fig. 1). Following most damages and injuries such as partial hepatectomy, the liver regenerates by self-replication of hepatocytes driven by dedifferentiation [11]. However, when hepatocyte proliferation is compromised, formation of duct-like oval cells (HPCs) with a mixed mesenchymal and epithelial expression signature has been observed [12]. These progenitor cells are assumed to originate from terminal branches of the intrahepatic biliary system. Seminal work has demonstrated that these cells can give rise to hepatocytes [13]. Recent studies using mouse models of chronic liver insults have indicated that new hepatocytes originate from preexisting hepatocytes rather than from distinct nonparenchymal stem cell populations [14]. Although there have been numerous reports regarding cellular origin of HCC, owing to the unavailability of comprehensive mouse models that can accurately mimic human liver architecture, molecular mechanisms involved in hepatocyte dedifferentiation or progenitor cells expansion remain to be elucidated.
CURRENT STATE OF HEPATOCELLULAR CARCINOMA THERAPEUTICS
Systemic treatments against liver cancer show poor efficacy mainly because of high resistance and poor response to therapeutics and the large degree of heterogeneity of liver tumors. Multiple treatment options are available for HCC, including curative resection, liver transplantation, radiofrequency ablation, trans-arterial chemoembolization, and systemic targeted agent like sorafenib [15,16]. Although progress has been made in combinational treatment for liver cancer, current therapies for HCC are only marginally effective. They often exacerbate underlying liver disease. The most important new trends in therapy against HCC are advances in molecular targeted therapy and employment of immunotherapy. In addition, targeted agents tailored to inhibit specific protein kinases involved in intracellular signal transduction pathways known to drive tumor progression and recurrence, including RAS/RAF-mitogen-activated protein kinase (MAPK) pathway, phosphatidylinositol 3-kinase (PI3K)/AKT pathway, WNT/β-catenin pathway, insulin-like growth factor (IGF), hepatocyte growth factor (HGF)/c-MET pathway, and growth factor-regulated angiogenic signaling pathways are being developed and tested in a variety of preclinical and clinical trials [17-22]. For instance, sorafenib is a recently developed oral multi-targeted inhibitor for RAF, platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF) receptors. It has been proven to be effective against advanced HCC [21-24]. Although the efficacy of sorafenib has been demonstrated, it is not considerably effective. It improves patient survival for a median of less than 3 months and delays radiological disease progression by an average of 2 months [23]. Moreover, response to sorafenib is highly variable among HCC patients owing to high heterogeneity of HCC. In addition, drug efficacy and mortality of HCC are various. They are not solely influenced by tumor stage [23,24]. These data strongly suggest that it is critical to identify novel targets and therapeutic strategies to efficiently treat HCC, particularly for cases that escape current treatment strategies and thereby show recurrence.
HCC shows high resistance and poor response to systemic treatments. Although progress has been made in combinational treatment of this disease, no effective disease-modifying therapies for HCC have been developed to date. An improved understanding of the mechanisms responsible for liver cancer initiation and progression will facilitate the development of new targeted therapies for liver cancer. However, identification of cellular origins of liver cancer remains a challenging issue. Therefore, a comprehensive understanding of HCC pathogenesis will enable rational identification of biomarkers and therapeutic targets and provide insights into mechanistic-based strategies for treatment of HCC. It is also evident that an understanding of signaling processes involved in hepatic dedifferentiation will provide clues to the pathogenesis of HCC development and the molecular mechanisms involved in self-renewal-related tissue repair in response to liver injury/damage. This is also critical in the search for molecular targets for therapeutics as specific pathways may be selectively targeted via pharmacological means to manipulate oncogenic dedifferention of hepatocytes. This has been partially proven to be feasible with imatinib mesylate, a tyrosine kinase inhibitor, that can inhibit liver progenitor cell (and also dedifferentiated hepatocyte) responses and liver cancer formation following chronic liver injury [2,3]. Alternatively, an experimental strategy to manipulate liver progenitor cell and dedifferentiated hepatocytes may be used to enhance liver regeneration after surgical resection.
UNIQUE IMMUNE RESPONSES DURING HCC PROGRESSION AND LIVER REGENERATION
The ability of immunotherapy to elicit nontoxic, systemic, long-lived anti-tumor activity makes it particularly well-suited for use in the setting of HCC. Although therapeutic benefit has been seen in early clinical trials, the efficacy of immune-based therapies is limited by several unique properties of HCC, most notably the inherently tolerogenic character of the liver in both healthy and diseased (chronically-infected or tumor-bearing) states [24]. To profoundly improve clinical outcomes for HCC patients by application of immunotherapy, therapeutic regimens that can counteract these immunosuppressive mechanisms and amplify tumor-specific immunity should be developed (Fig. 2). The most formidable barrier to immune-based therapy for HCC might be the unique immunobiology of the liver. Liver tissue is routinely exposed to damaging agents through the bloodstream. It captures and eliminates toxins and microbial products arising from arterial circulation and intestines. In response to continual pathogen exposure, the liver has evolved a unique and redundant system of immune regulation demonstrated by relatively low rates of liver allograft rejection and limited need for immune suppression after transplantation [24]. Hepatocytes contribute to the liver’s inherent tolerogenicity by priming naïve T cells in the absence of co-stimulation, resulting in defective cytotoxicity and clonal deletion [25]. Otherwise, naïve T cells that initially encounter antigens in the lymph nodes acquire full effector function, suggesting that effective immunotherapy in the setting of HCC must avoid intrahepatic T cell priming (Fig. 2) [15,26].

Regulation of hepatocarcinogenesis by tumor immunity. HCC, hepatocellular carcinoma; NASH, non-alcohol steatohepatitis; PD-1, programmed cell death protein 1; DC, dendritic cell; NK, natural killer cell; CTL, cytotoxic T lymphocyte; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell; Th1, type 1 T helper cell; Th2, type 2 T helper cell; Th17, T helper 17 cell.
Notably, T cell exhaustion often occurs during chronic infection and prevents optimal viral control. Molecular pathways involved in T cell exhaustion remain poorly understood. After acute viral infection, memory CD8+ T cells rapidly reactivate effector functions. With a high degree of proliferative potential, memory CD8+ T cells are maintained by antigen-independent homeostatic proliferation [15,26,27]. These properties allow memory T cells to confer protective immunity. In contrast, during chronic viral infection, antigen-specific CD8+ T cells initially acquire effector functions but gradually become less functional as the infection progresses (Fig. 2) [15,26,28]. Persistent hepatitis B virus (HBV) or hepatitis C virus (HCV) infection can contribute to the development of HCC by driving chronic liver inflammation which might trigger hepatic dedifferentiation. Chronic HBV or HCV infections have strong immunosuppressive properties. In individuals with persistent HBV infection, programmed cell death protein 1 (PD-1) expression by peripheral blood CD8+ cytotoxic T lymphocytes is upregulated when disease progresses from cirrhosis to HCC [29,30]. Dysfunctional T cell responses are also observed in chronic HBV- or HCV-infected patients [31]. Furthermore, the frequency of intrahepatic regulatory T cell (Treg) in these patients is correlated with viral load, suggesting that Treg accumulation in a chronically-infected liver may abrogate CD8+ T cell-mediated clearance of these viruses [30,32]. These tolerogenic factors may accumulate during HBV/HCV-mediated hepatocarcinogenesis and coincide in advanced HCC lesions, facilitating an aggressive and effective counterattack to anti-HCC immunity. Because the strategy to overcome HCC-mediated immunosuppression involves direct reactivation of hyporesponsive tumor-specific T cells, a not-yet-attempted therapeutic option as a regimen to reactivate T cell function will be important.
CONCLUSION
It is evident that hepatic dedifferentiation in response to various types of liver injury/damage is a substantial process involved in tissue repair. It acts in opposition to HCC development. However, molecular mechanisms responsible for reprogramming fully-differentiated hepatocytes into a dedifferentiated state that is plastic and reversible remain unclear. Molecular signaling molecules that can discriminate between liver regeneration and oncogenic proliferation remain a mystery. This reviewer may improve our understanding of the biology of liver cancer with pivotal implications from a therapeutic perspective.
Notes
No potential conflict of interestrelevantto this article was reported.
Acknowledgements
This study was supported by grants (2017R1A2B3006776 & 2017M2A2A7A01070267) of National Research Foundation (NRF) funded by the Ministry of Education, Science, and Technology (MEST), Republic of Korea.