Intrafamilial variability and clinical heterogeneity in a family with PLA2G6-associated neurodegeneration
Article information

Abstract
Phospholipase A2 group VI (PLA2G6)-associated neurodegeneration (PLAN) is an autosomal recessive neurodegenerative disease with a wide clinical spectrum; however, the genotype-phenotype correlation is unknown. Here, we report different phenotypes in one family with the same genotype. A 28-year-old male presented with slowly progressive gait disturbance with spasticity. Onset occurred at 11 years. Interestingly, his younger brother, a 24-year-old male, presented with progressive Parkinsonism, which began at 22 years. He showed excellent response to levodopa but developed a fluctuating medication response and levodopa-induced dyskinesia 1 year after starting levodopa medication. He also demonstrated hyperreflexia, but no spasticity. Dopamine transporter imaging showed reduced uptake in the bilateral putamen. In whole-exome sequencing and Sanger sequencing, a homozygous pathogenic variant (p. R747W) in the PLA2G6 gene was detected in both cases. Despite different clinical features, both subjects had hyperreflexia during the examination and claval hypertrophy was shown on the brain magnetic resonance imaging.
INTRODUCTION
Phospholipase A2 group VI (PLA2G6)-associated neurodegeneration (PLAN) is an autosomal recessive neurodegenerative disease with multiple phenotypes, including infantile neuroaxonal dystrophy, neurodegeneration with brain iron accumulation, and dystonia-Parkinsonism [1,2]. The clinical spectrum of PLAN has been proposed to include various phenotypes associated with PLA2G6 mutations. Although various phenotypes are known in patients with PLAN, genotype-phenotype correlation has yet to be clearly elucidated. Further, there are no reports demonstrating various phenotypes even in one family with the same genotype. Here, we describe a family with the same genotype of PLAN, but with phenotypic variability.
CASE REPORTS
Subject 1
A 28-year-old male with progressive spastic gait disturbance that began at the age of 11 presented at Samsung Medical Center, Seoul, Korea. He was born to healthy consanguineous parents of Arabian ancestry. He had no history of preterm delivery or perinatal injury, and his developmental milestones were normal. There was no family history of movement disorders, except in his younger brother (Subject 2). The familial pedigree is illustrated in Fig. 1. He was normal in cognitive and language skills and finished his master’s degree in college. The only accompanying symptom was urinary urgency and incontinence.
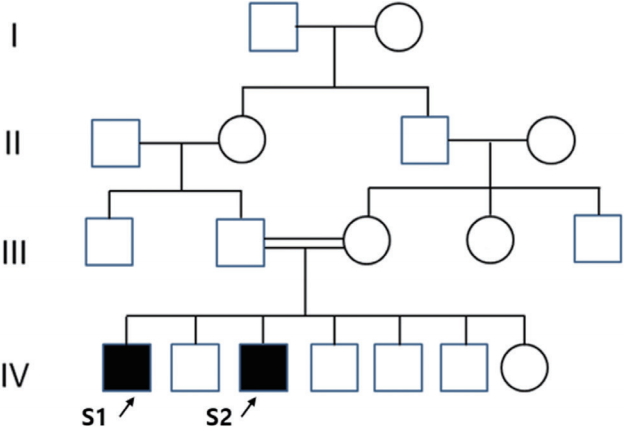
Pedigree of the index subjects: black symbols indicate affected individuals and arrows indicate subjects.
When he came to our clinic, spasticity was present in the hip adductor muscles, as well as in the knee and ankle joints. Sensory disturbance of lower limbs was absent. Brisk reflexes were present on both limbs, and pathologic reflexes, such as Hoffman sign, ankle clonus and Babinski sign, were also present. Grade 4 mild muscle weakness was present with both ankle dorsiflexions. He demonstrated spastic gait with dystonic ankle movements interfering speed of gait, but no rigidity or bradykinesia. A nerve conduction study on lower limbs was normal. There was no iron accumulation, but claval hypertrophy was shown on the brain magnetic resonance imaging (MRI) (Fig. 2A). The cervical through thoracic spinal MRI was normal.

(A) T2 fluid attenuated inversion recovery (FLAIR) midsagittal section image of subject 1 shows claval hypertrophy (white arrow). (B) T2 FLAIR midsagittal section image of subject 2 shows claval hypertrophy (white arrow) and a vertical corpus callosum (black arrow). (C) Dopamine transporter image of subject 2 demonstrated significantly reduced uptake in the bilateral putamen.
Whole exome sequencing for subject 1 revealed a homozygous c.2239C>T (p.R747W) pathogenic variantin thePLA2G6 gene. Sanger sequencing showed that both of the parents were heterozygous for this variant (Fig. 3). His gait disturbance did notrespond with levodopa treatment but partially improved with baclofen (30 mg/day) and botulinum toxin type A injection on the tibialis posterior muscle (50 units, each).
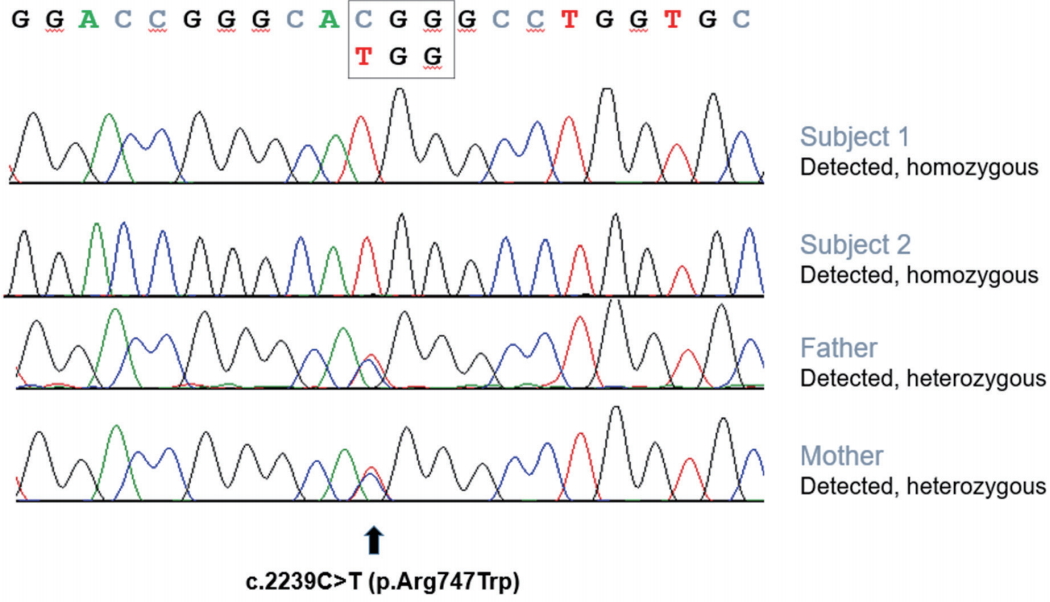
Sequencing analysis of index subjects showing homozygous variants in phospholipase A2 group VI (PLA2G6). Analysis of parental samples demonstrated that the PLA2G6 variants were heterozygous, which is consistent with autosomal recessive inheritance.
Subject 2
The younger brother of subject 1, a 24-year-old male, presented at our clinic with slowly progressive Parkinsonism that began at the age of 22. Like subject 1, he had no history of preterm delivery or perinatal injury, and the developmental milestones were normal. Urinary urgency was combined, while general cognition was normal.
On the neurologic exam, bradykinesia and rigidity were present in both limbs, but more prominently on the left side. Additionally, rest tremor was seen in both arms. Deep tendon reflexes were brisk and the Babinski sign was present in both lower limbs. F-18 fluorinated-N-3-fluoropropyl-2-b-carboxymethoxy-3-b-(4-iodophenyl)nortropane positron emission tomography (FP-CIT PET) demonstrated significantly reduced uptake in the bilateral putamen (Fig. 2C). Additionally, corpus callosum changes and claval hypertrophy were seen on the brain MRI (Fig. 2B).
We performed Sanger sequencing of the PLA2G6 gene, and the same homozygous pathogenic variant (p.R747W) was detected (Fig. 3). His Parkinsonism improved dramatically with levodopa medication, but he developed wearing-off phenomenon and levodopa-induced dyskinesia within 1 yearfrom the levodopa start date.
DISCUSSION
Here, we described two different phenotypes of the same genotype of PLAN in one family. One phenotype was levodopa-responsive Parkinsonism and the other was spastic paraplegia. Both shared hyperreflexia on lowerlimbs and pyramidal tract syndrome, but totally different main phenotypic presentations in the same genotype are rarely reported. Interestingly, PLAN covers various phenotypes of PLA2G6 mutation, and various phenotypes of PLAN are described according to the age of onset (e.g., infantile neuroaxonal dystrophy occurs in childhood and dystonia-Parkinsonism occurs in early adulthood). However, even though various phenotypes of PLAN have been reported, there are currently insufficient studies on phenotype-genotype correlation. Further, our patients were similar in age but presented with completely different phenotypes in a single family with the same genotype, unlike various phenotypes in PLAN. To the best of our knowledge, this is the first report of intrafamilial variability in monogenic PLAN. The mechanisms responsible for determination of phenotypes are not yet known.
The first phenotype of our subjects was levodopa-responsive Parkinsonism. Parkinsonism is a commonly reported symptom in PLAN patients, and the same genotype (p. R747W) has been previously reported in a patient with levodopa-responsive Parkinsonism [3,4]. In accordance with previously reported PLAN cases with Parkinsonism, our case also showed normal birth, achieved normal milestones, and symptoms began in adulthood. All cases progressed rapidly afterthe onset and showed a dramatic response to levodopa therapy, but an impulse control disorder or early dyskinesia appeared. Additionally, our case also revealed presynaptic dopaminergic loss by FP-CIT PET imaging study.
In terms of spastic paraplegia, 60 hereditary spastic paraplegia (HSP)-related genes have been reported, but the underlying causes of HSP still remain unidentified [5]. Recently, PLAN with phenotype of complicated HSP was reported [6,7], but pure form spastic paraplegia was not reported. With whole exome sequencing, we excluded associated genes for HSP in subject 1; thus, we suggest pure form spastic paraplegia is a new phenotype of PLAN based on our case.
Despite different main phenotypes of our patients, both subjects also demonstrated common clinical features. First, pyramidal tract syndrome was seen in both subjects. Additionally, both cases had claval hypertrophy (Fig. 2A, B). In terms of brain imaging, abnormal iron accumulation is considered as a characteristic sign of neurodegeneration with brain iron accumulation, but iron accumulation is no longer a pathognomonic finding in PLA2G6 mutations. Similarly, our cases did not show iron accumulation in brain. Additionally, various MRI findings, including cerebral and/or cerebellar atrophy, corpus callosum changes, or claval hypertrophy, could be observed in patients with PLAN [8]. Subject 2 showed corpus callosum changes as well as claval hypertrophy (Fig. 2B). Therefore, pyramidaltract syndrome and claval hypertrophy could be the diagnostic clue for PLAN to overcome the phenotypic diversity.
In conclusion, the affected patients of PLA2G6 mutation showed clinical heterogeneity within a family.PLA2G6 mutation can present as a pure form of spastic paraplegia or L-dopa responsive Parkinsonism with motor fluctuation. Additionally, we describe a previously unrecognized phenotype ofPLA2G6 mutations thatis pure form spastic paraplegia.
Notes
No potential conflict of interestrelevantto this article was reported.