INTRODUCTION
Frontotemporal lobar dementia (FTD) syndromes include a behavioral variant (bvFTD) and two subtypes of primary progressive aphasia [1,2]. These syndromes are united by overlapping clinical and anatomical features and their link to the underlying frontotemporal lobar degeneration pathology (FTLD) [1,2]. FTLD is characterized by neuronal and glial inclusions immunoreactive for hyperphosphorylated tau (FTLD-tau), or TAR DNA-binding protein of 43 kDa (TDP-43, FTLD-TDP) [3,4]. Despite attempts to associate FTD clinical syndromes with specific FTLD neuropathological diagnoses, no such correlation has yet been proven invariant [5-7]. In particular, non-fluent variant primary progressive aphasia (nfvPPA) has been proven to have various pathological diagnoses. Over 60% of nfvPPA cases show FTLD-tau, including Pick’s disease (PiD), progressive supranuclear palsy (PSP), and corticobasal degeneration. Most of the remaining cases show TDP type A and Alzheimer’s disease pathology [6,8]. Herein, we report two patients clinically diagnosed with nfvPPA that displayed different pathological findings at autopsy.
CASE REPORTS
The collection of the data was conducted as set forth in the Declaration of Helsinki. This study was approved by the Institutional Review Board of Samsung Medical (IRB Rile No. 2016-11-111) and we obtained informed consent from the participant and their next of kin (daughter and spouse).
Case 1
A 63-year-old right-handed woman visited our clinic due to language dysfunction and personality change. She had started to show effortful, non-fluent speech and a decreased amount of speech 1 year prior to her first visit. After 1 year, she showed abulia and blankly watched TV all day. She occasionally got upset for no reason. There was no familial history of dementia. She had hypertension and hyperlipidemia, and was on hemodialysis for chronic renal failure.
On neurological examination, the patient showed motor impersistence and grasp reflex on the bilateral upper extremities. She scored 22 out of 30 on the Mini-Mental State Examination (MMSE). There were no signs of Parkinsonism. Detailed neuropsychological tests revealed that her attention, language, and verbal memory were decreased; especially her frontal executive function, which was severely decreased. On the Western Aphasia Battery (WAB), she demonstrated non-fluent speech and a lack of verbal response. The average number of word segments per utterance was one; word comprehension was mildly decreased, but complex sentence comprehension was remarkably decreased, confrontational naming ability was severely decreased, and repetition was mildly decreased. In particular, she showed word-finding difficulty and delayed word retrieval. There was no effortful speech, agrammatism, or dysarthria. These results indicated a moderate degree of Broca’s aphasia (Table 1). Her fluid-attenuated inversion recovery (FLAIR) images showed bilateral frontal and anterior temporal atrophy (Fig. 1). Based on her clinical symptoms, neuropsychological and language tests, and brain magnetic resonance imaging (MRI), the patient was clinically diagnosed with nfvPPA. One year after her first visit, she displayed blunted affect and did not welcome her children. Two years after her first visit, her spontaneous speech was limited to one word. She was not able to wash herself, making activities of daily living impossible without aid. Four years after her first visit, she was found dead at home. After her death, the patient’s body underwent a brain autopsy at the Samsung Medical Center.
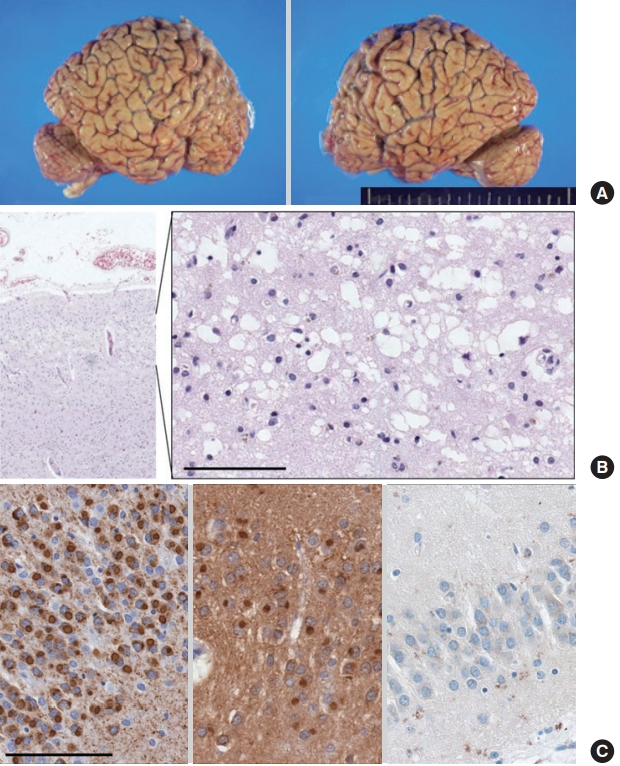
Postmortem examination showed a decreased brain weight of 988 g (standard for age: 1,360 g). Gross examination revealed moderate to severe gyral atrophy in the bilateral frontal lobes, and mild gyral atrophy in the bilateral parietal and medial temporal lobes (Fig. 2A). Histological examination revealed microvacuolation with severe neuronal loss and gliosis in the frontal cortex layers 2 and 3 (Fig. 2B). Mild neuronal loss and gliosis were present in the parietal cortex layer 2, temporal cortex layer 2, amygdala, basal nucleus, basal ganglia, and dentate nucleus. Tau-positive intraneuronal cytoplasmic inclusions (Pick bodies), identified by tau and 3-repeat tau stains, were located in the frontal, parietal, and temporal cortex layers. Pick bodies were also found in the dentate gyrus, hippocampus, subiculum, basal ganglia, thalamus, periaqueductal gray matter, locus coeruleus, and pontine nucleus (Fig. 2C). In the absence of TDP-43 pathology and α-synucleinopathy, a pathological diagnosis of FTLD-tau, PiD, was rendered.
Case 2
A 71-year-old right-handed man visited our clinic because of language dysfunction, left hand clumsiness, and memory impairment. He had started to show slow speech velocity, stuttering, and word-finding difficulty half a year prior to his first visit. There was no familial history of dementia. He was suffering from hypertension.
On neurological examination, the patient showed ideomotor apraxia and poor performance in fist-edge-palm. He also experienced bradykinesia in the left upper limb. He scored 25 out of 30 on MMSE. Detailed neuropsychological tests revealed that his attention, language, verbal memory, and frontal executive function, except visuospatial function and visual memory, were decreased. On WAB, he was overall fluent, but showed frequent hesitation, halting, repetition of syllables, phonemic paraphasia, and word-finding difficulty. Word comprehension, repetition, and naming were slightly decreased. These results indicate a mild degree of anomic aphasia (Table 1). His apolipoprotein E genotype was e3/e3. On FLAIR images, diffuse brain atrophy and periventricular white matter hyperintensities in the anterior and posterior portions were seen. On gradient echo images, strictly multiple lobar microbleeds and cortical superficial siderosis in the left parietal cortex were observed, suggesting probable cerebral amyloid angiopathy (CAA). [18F]-fluorodeoxyglucose positron emission tomography (FDG-PET) revealed moderate hypometabolism in the bilateral frontal cortex, which was more severe on the left side (Fig. 3). Based on his clinical symptoms, WAB, detailed neuropsychological tests, brain MRI, and FDG-PET, the patient was clinically diagnosed with nfvPPA. Three years after his first visit, he showed cogwheel rigidity in the bilateral upper and lower limbs. However, the patient did not show axial rigidity, postural instability and vertical gaze limitation. The overall language function worsened, and he demonstrated phonemic paraphasia, neurogenic stuttering, and perseveration. He did not show dysarthria or apraxia of speech. These results indicate a moderate degree of anomic aphasia (Table 1). Four years after his first visit, he showed impatience and repetitive motor behaviors, such as tearing papers. Six years after his first visit, he was unable to walk anymore and died of pneumonia at the age of 77 years. After his death, his body underwent a brain autopsy at the Samsung Medical Center.
Postmortem examination showed a decreased brain weight of 940 g (standard for age: 1,300 g). Gross examination revealed marked atrophy of the bilateral precentral gyrus with moderate atrophy of the bilateral frontal and superior temporal gyri (Fig. 3A). On microscopic examination, tufted astrocytes were identified in the middle frontal gyrus, inferior parietal lobule, and occipital lobe. Globoid tangles and pretangles were observed in the temporal lobe, putamen, subthalamic nucleus, pons, and medulla oblongata (Fig. 3C). β -Amyloid plaques with amyloid angiopathy were present in the occipital cortex, amygdala, hippocampus, and cerebellum. Atherosclerosis in the vessels of the circle of Willis and hemosiderin pigmentation in the left inferior parietal lobule, suggestive of old subarachnoid hemorrhage, were noted. Microinfarcts were identified in the left frontal, temporal, and parietal lobes. Overall, the patient was diagnosed with FTLD-tau, PSP with Alzheimer’s disease (Braak Stage II), CAA, and cerebrovascualr disease.
DISCUSSION
Here, we report two clinically diagnosed nfvPPA cases. Both patients showed decreased fluency and effortful and decreased speech. As the disease progressed, they showed not only language dysfunction but also other symptoms, such as behavioral changes or motor dysfunction. However, they demonstrated showed several distinguished characteristics according to the pathological diagnosis. The patient pathologically diagnosed with PiD showed early age at onset and initial personality change accompanied by language dysfunction. On the other hand, the patient with PSP showed late age at onset and initial parkinsonian features accompanied by language dysfunction.
Patients with nfvPPA might show distinct clinical features depending on the underlying pathologies. Clinicopathological association between nfvPPA clinical syndrome and FTLD neuropathological diagnosis is not specific; nfvPPA is associated with multiple pathologies, including tauopathy, TDP type A pathology, and Alzheimer’s disease pathology [6,8]. The first case was pathologically diagnosed as PiD. PiD, a rare subtype of FTLD-tau, is characterized by right dominant frontotemporal atrophy and clinical features of bvFTD. However, a few patients with PiD have showed left dominant frontal and peri-Sylvian atrophy and clinical features of nfvPPA. Gross neuropathological findings of PiD show circumscribed lobar atrophy, and microscopic findings are characterized by neuronal loss, gliosis, and argyrophilic and round intraneuronal inclusions (Pick’s body). Pick’s body is composed of a tau protein containing three microtubule-binding repeats (3R) [9].
PSP primarily presents with atypical parkinsonism with axial rigidity, frequent falls, postural instability, and progressive gaze palsy [9]. In particular, 80% to 90% of clinically suspected progressive supranuclear palsy syndrome (PSPS) show PSP pathology. However, approximately 10% of PSP presents the clinical features of nfvPPA, similar to our second case [6]. The cardinal neuropathological findings consisted of tufted astrocytes, globose neurofibrillary tangles, and oligodendroglial coiled bodies. These astrocytic, neuronal, and oligodendroglial lesions of PSP are mainly composed of tau protein containing four microtubule-binding repeats (4R) [9].
In conclusion, clinically diagnosed nfvPPA patients are expected to have various pathological diagnoses. Patients with nfvPPA might show distinct clinical features depending on the underlying pathologies.